Скачать презентацию
1
Подготовила: студентка 2 группы 3 мед. ф-та 5 курса Малько Т.Г.

2
Фенилкетонурия Это наследственная аминоацидопатия, связанная с нарушением метаболизма фенилаланина, в результате мутационной блокады ферментов приводящая к стойкой хронической интоксикации и поражению ЦНС c выраженным снижением интеллекта.

3
Этиология и патогенез В результате мутации гена, контролирующего синтез фенилаланингидроксилазы, развивается метаболический блок на этапе превращения фенилаланина в тирозин, вследствие чего основным путем преобразования фенилаланина становится дезаминирование и синтез токсических производных - фенилпировиноградной, фенил-молочной и фенилуксусной кислот..

4
В крови и тканях значительно увеличивается содержание фенилаланина (до 0,2 г/л и более при норме 0,01-0,02 г/л) Заболевание наследуется по аутосомно- рецессивному типу
 Заболевание наследуется по аутосомно- рецессивному типу")
5
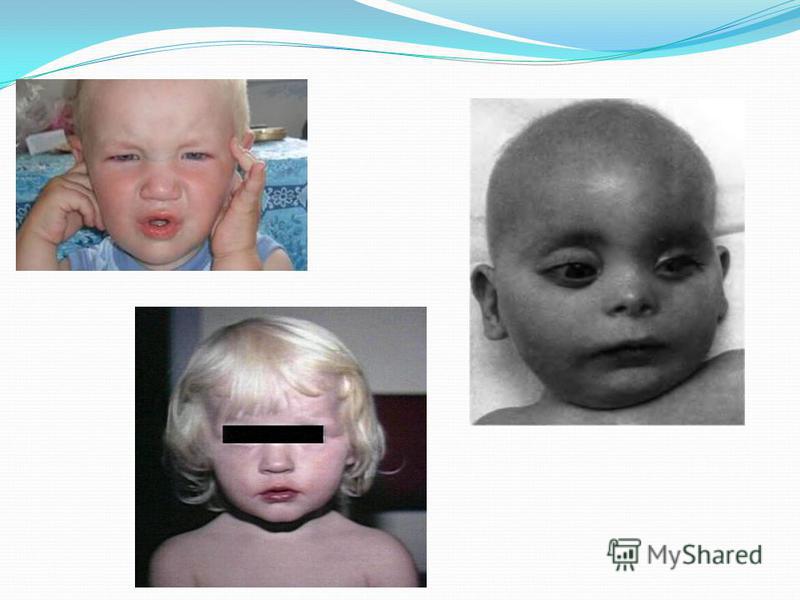
Симптомы Появляются в возрасте 2–6 месяцев. повышенная возбудимость ребенка; умственная отсталость; специфическая походка; необычное положение конечностей; специфическая осанка, поза при сидении; стереотипные движения; судороги; повышение сухожильных рефлексов; микроцефалия; изменения кожи: гипопигментация, сухость, экзема, склеродермия; светлые радужки, катаракта; волосы гипопигментированы; специфический мышиный запах тела.

7
Дефект познавательной деятельности Слабое развитие интроспекции Невозможность сосредоточения внимания Полное отсутствие математических способностей Рисуют плохо Инертны Развита способность к индукции Грамматический строй и связная речь имеют сильные нарушения Плохая координация Мелкая моторика рук развивается с большим трудом Ловкость ребёнка снижена или отсутствует

9
Формы Классическая фенилкетонурия. При этой форме болезнь наследуется как рецессивный признак. При отсутствии лечения больные не доживают до 30 лет.Частота распространения данной формы заболевания – 1 ребенок на новорожденных. Вариантная фенилкетонурия. Такая форма не является наследственным заболеванием, она возникает из-за мутации в генах. Встречается она реже: всего один случай заболевания на миллион новорожденных детей. Клинические проявления данной формы схожи с классической фенилкетонурией, однако протекает она тяжелее. Данная форма также называется «злокачественной фенилкетонурией», характеризуется тяжелыми неврологическими нарушениями и ранней смертью.

10
Клинические проявления злокачественной фенилкетонурии Тяжелые и быстро прогрессирующие неврологические нарушения Гипотония Спастический синдром Атаксия Нарушение глотания Беспокойство Прогрессирующая умственная отсталость Тяжело купирующиеся приступы судорог

11
Виды ФКУ Фенилкетонурия I (классическая фенилкетонурия). Эта форма болезни является наследственным заболеванием (аутосомно-рецессивным), которое возникает при мутации гена фенилаланингидроксилазы. Фенилкетонурия II (атипичная фенилкетонурия) – недостаточность фермента дегидроптеринредуктазы, которая приводит к нарушениям процессов восстановления тетрагидробиоптерина. В результате возникают метаболические блоки в процессе превращения фенилаланина в тирозин. Фенилкетонурия III – недостаточность фермента 6-пирувоилтетрагидроптеринсинтазы, который участвует в процессах синтеза тетрагидробиоптерина из дигидронеоптеринтрифосфата, что приводит к нарушениям, сходным с расстройствами при фенилкетонурии II. Материнская фенилкетонурия – снижение уровня интеллекта у детей женщин, болеющих фенилкетонурией.
. Эта форма болезни является наследственным заболеванием (аутосомно-рецессивным), которое возникает при мутации гена фенилаланингидроксилазы. Фенилкетонурия II (атипичная фенилкетонурия) – недост")
13
Диагностика Диагноз фенилкетонурии необходимо поставить сразу при рождении, для профилактики тяжелых последствий ФКУ. Вот поэтому в роддоме на 4-й -5-й день жизни новорождённого(доношенного) и на 7-й день- у недоношенного берут кровь на обследование на ФКУ. Через час после кормления каплей капиллярной крови пропитывают специальный бумажный бланк. При концентрации фенилаланина в образце крови более 2,2 мг% ребенка с родителями направляют в медико- генетический центр для осмотра, обследования, уточнения диагноза.
 и на 7-й день- у недоношенного берут кровь на обследование")
14
. Уровни ФА в скрининге: 3-8 мг% - родителей или опекунов информируют о необходимости выполнения контрольного исследования (колориметрическим методом). Если в контрольном исследовании уровень ФА выше 2 мг%, то необходимо оповестить родителей/опекунов и пригласить в письменной форме в специализированный диагностический лечебный центр; выше 8 мг% - необходимо оповестить родителей/опекунов о результате скринингового теста и пригласить в письменной форме в специализированный диагностический лечебный центр. Во время первого визита врач- специалист (чаще всего педиатр или генетик) обязан предоставить родителям/опекунам исчерпывающую информацию относительно предполагаемого заболевания, его причин и возможностей лечения.педиатр В связи с существованием ряда причин появления гиперфенилаланемии (фенилкетонурия, нетипичные формы фенилкетонурии и тирозенемия), необходимо провести дифференциальную диагностику. Если подтвердится дефицит гидроксилазы фенилаланина (фенилкетонурия), применяется диетотерапия. В случае, если причиной окажется нарушение обмена веществ иного рода, будут избраны иные методы лечения
. Если в контрольном исследовании уровень ФА выше 2 мг%, то необходимо оповестить родителей/опекунов")
15
Лечение Единственным эффективным методом лечения больных ФКУ является специализированная диетотерапия с момента установления диагноза. Диета при ФКУ - это: Уменьшение дозы фенилаланина согласно индивидуальной толерантности фенилаланина, что означает уменьшение дозы натурального белка в суточном рационе Обеспечение соответствующей для нормального развития дозы белка (дополнительный белок без фенилаланина) из продуктов лечебного питания ФКУ Обеспечение соответствующей дозы энергии с использованием специальных низкобелковых продуктов Обеспечение соответствующей дозы витаминов, макро- и микроэлементов – главным образом из препаратов ФКУ и других источников.

16
Продукты питания ЗАПРЕЩЕНЫ: Яйца Рыба Мясо и мясные продукты (мясные и колбасные изделия) Домашняя птица Зерновые продукты: хлебобулочные изделия, мука, каши, хлопья, макароны, кондитерские изделия Стручковые растения: фасоль, горох, соя Семена: кукуруза, мак, льняное семя Орехи Шоколад Молочные продукты: сыр, йогурт, творог, сметана, мороженое Желатин Аспартам
 Домашняя птица Зерновые продукты: хлебобулочные изделия, мука, каши, хлопья, макароны, кондитерские изделия Стручковые растения: фасоль, горох, соя Семена: куку")
17
Продукты питания: Разрешены в строго ограниченном количестве Овощи Картофель Фрукты Рис Джемы, варенья Мёд Масло Mаргарин Щербеты Хлебобулочные изделия с низким содержанием белка Макароны и мучные продукты, изготовленные из муки с низким содержанием фенилаланина

18
Продукты питания Разрешены в неограниченном количестве Сахар Растительные масла Минеральная вода Чай Фруктовые конфеты Леденцы Загустители (караген, пектина, гуаровая

19
Питание Для детей первого года лечебные продукты приближены по составу к грудному молоку. Это – смеси «Афенилак », «Лофенилак» и др. Для детей старше года такие смеси, как «Фенил-фри», «Максамум-ХР», «Тетрафен» и др. Смесь «Максамум-ХР» назначается детям старшего возраста(после 6-ти- 8-ми лет), а также беременным женщинам, страдающим ФКУ. Контроль за содержанием фенилаланина в сыворотке крови проводят в начале лечения – еженедельно, а затем при нормализации показателей – ежемесячно в течении первого года жизни, у детей старше года при нормальных показателях – 1 раз в 2-3 месяца.

20
Профилактика Развитие фенилкетонурии профилактировать нельзя (наличие дефектного гена в организме ребенка предотвратить невозможно). Для определения риска рождения ребенка с фенилкетонурией проводят предварительные генетические консультации, которые рекомендуются для супружеских пар, состоящих в кровнородственном браке, уже имеющих больного ребенка, имеющих родственников с данным заболеванием. Риск рождения у родителей-носителей дефектного гена ребенка с фенилкетонурией составляет 1:4. При развившемся заболевании имеет значение только профилактика развития тяжелых повреждений мозга путем своевременного назначения диеты. Для пренатальной диагностики (до родов) возможно проведение генетического анализа (исследование генов родителей) в медико- генетической консультации для предсказания возможного развития заболевания у ребенка (проводится в семьях, где уже рождались дети с фенилкетонурией).
. Для определения риска рождения ребенка с фенилкетонурией проводят предварительные генетические консультации, которые")
21
Спасибо за внимание!













 Заболевание, связанное с интоксикацией фтором, возникающее в результате повышенного содержания фтора в питьевой воде.")










