Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
Проведение исследований дженериков на биоэквивалентность в рамках формирующейся регуляторной платформы Государственный экспертный центр МЗ Украины Директор департамента доклинических и клинических исследований к.м.н. Николаева В.В.

2
Страны с жесткой регуляторной политикой США год Япония год до нашей эры Соединенное Королевство Великобритании и Северной Ирландии год Италия год Французская Республика год (Пятая Республика) Страны ЕС, например:
 Страны ЕС, например:")
3
Страны СНГ

4
Ukraine областей Население около 45 млн Четкое законодательство: Конституция, Законы, Постановления КМ, Приказы Более чем опытных Сlinical Trials Sites

5
Международные регламенты оценки биоэквивалентности EC, EMA CPMP/EWP/QWP/1401/98/ Rev.1 Guideline on the Investigation of Bioequivalence, ЕМА-2010 ВОЗ WHO Technical Report Series, No. 937 США (FDA) Guidance for Industry. Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based an a Biopharmaceutics Classification System, 2000 Japan Guideline for Bioequivalence Studies for Different Strengths of Oral Solid Dosage Forms, Japan,
 Guidance for Industry. Waiver of In Vivo Bioavailabil")
6
Международные регламенты оценки биоэквивалентности EC, EMA ВОЗ Japan GCP GLP GMP Хельсинская декларация США (FDA) GSPGRP
 GSPGRP")
7
7 Соотношение рынка оригинальных и генерических препаратов (Аптека. – 2010 (755) )
. - 34.)")
8
ГЕНЕРИЧЕСКИЙ ПРЕПАРАТ лекарственный препарат, используемый в медицинской практике взаимозаменяемо с инновационным препаратом, производящийся, как правило, без лицензии от компании разработчика и реализуемый после истечения срока действия патента или других исключительных прав ИННОВАЦИОННЫЙ ПРЕПАРАТ патентованный препарат, впервые получивший разрешение на маркетинг на основании документов, подтверждающих его эффективность, безопасность, качество, в соответствии с требованиями установленными на момент получения разрешения

9
9 Сходство Субстанция Доза Лекарственная форма Сходство Субстанция Доза Лекарственная форма Различия Вспомогательные вещества Технология производства Различия Вспомогательные вещества Технология производства Биоэквивалентность Равная биодоступность оригинальный препарат VS генерик

10
Лекарственные препараты фармацевтически эквивалентны, если они содержат такое же количество того же действующего вещества (тех же действующих веществ) в тех же лекарственных формах, которые отвечают требованиям тех же или сопоставимых стандартов. (п Приказ МЗ Украины 191 от г., раздел ІІ приказа МЗ Украины 426 от г. в редакции приказа 3 от г.) два лекарственных средства биоэквивалентны, если они фармацевтически эквивалентны или фармацевтически альтернативны, и если их биодоступности после введения в одной и той же молярной дозе подобны в такой степени, что эффекты этих лекарственных средств по эффективности и безопасности будут оп сути одинаковыми. (Приказы МЗ Украины 690 от г. (с изменениями, внесенными приказом МЗ от ), 191 от г. и 426 от г. в редакции приказа 3 от г. ) Биоэквивалентность - Лекарственные препараты фармацевтически альтернативны, если они содержат тот же активный компонент, но отличаются по его химической форме (соль, эфир и т.д.) или лекарственной форме, или силе действия (п Приказа МЗ Украины 191 от г., раздел ІІ приказа МЗ Украины 426 от г. в редакции приказа 3 от г.) Биодоступность - скорость и степень, с которыми действующее вещество или его активный компонент абсорбируется (всасывается) из лекарственной формы и становится доступным в месте действия (Приказы МЗ Украины 690 от г. (с изменениями, внесенными приказом МЗ от ), 191 от г. и 426 от г. в редакции приказа 3 от г.).
 в тех же лекарственных формах, которые отвечают требованиям тех же или сопоставимых стандартов. (п.")
11
КИ по оценке биоэквивалентности ЛС с фармакокинетическими конечными точками является одним из методов доказательства терапевтической эквивалентности генерика к референтному препарату Лекарственный препарат терапевтически эквивалентен другому препарату, если он содержит то же действующее вещество или его терапевтически активную часть и клинически проявляет такую же эффективность и безопасность, как и препарат, эффективность и безопасность которого установлены" (п Приказа МЗ Украины 191 от г.) «Два ЛС считаются терапевтически эквивалентны, если они фармацевтически эквивалентны или фармацевтически альтернативны и после введения пациентам одним и тем же путем, в одной молярной дозе их безопасность и эффективность по сути аналогичны» (Приказ МЗ Украины 426 от г. в редакции приказа МЗ Украины 3 от г) Эквивалентность, или альтернативность за фармацевтическими характеристиками не обязательно предполагает терапевтическую эквивалентность, так как разница в наполнителях и / или производственном процессе и некоторых других переменных может привести к разному действию препарата. Фармацевтически эквивалентные ЛС, фармацевтически альтернативные ЛС могут быть или могут не быть биоэквивалентными / терапевтически эквивалентными.

12
Нормативные документы, которые регламентируют проведение исследований по оценке биоэквивалентности в Украине Закон Украины «О лекарственных средствах», (1996) ст.7, 8, 12 с изменениями, внесенными Законом Украины «О внесении изменений в некоторые законодательные акты Украины относительно клинических исследований лекарственных средств» от VI
 ст.7, 8, 12 с изменениями, внесенными Законом Украины «О внесении изменений в некоторые зак")
13
Порядок устанавливает основные требования к проведению КИ лекарственных средств, которые могут проводится на пациентах(добровольцах) по полной или сокращенной программе, в том числе к исследованиям биодоступности/биоэквивалентности, а также международным многоцентровым КИ (раздел І, п.2) CPMP/ICH/135/95 (E 6) Note for Guidance on Good Clinical Practice, Директива 2001/20/ЕС, Хельсинская декларация Приказ МЗ Украины от c изменениями, внесенными приказом МЗ Украины от Порядок проведения КИ лекарственных средств и экспертизы материалов КИ
 по полной или сокращенной программе, в том числе к исследованиям биодоступности/биоэквивалентности, а также междунаро")
14
Приказ МЗ Украины от Надлежащая клиническая практика (GCP) Руководство :2008 Приказ МЗ от в редакции приказа МЗ Украины от Раздел ІІІ, п.3.7 Порядка: «Планирование, проведение и отчетность всех фаз клинических исследований, в том числе исследований биодоступности / биоэквивалентности, осуществляются с соблюдением требований Руководства "Лекарственные средства. Надлежащая клиническая практика. СТ-Н МЗУ :2008"» CPMP/ICH/135/95 (E 6) Note for Guidance on Good Clinical Practice (Директива 2001/20/ЕС)
 Руководство 42-7.0:2008 Приказ МЗ от 23.09.09 690 в редакции приказа МЗ Украины от 12.07.2012 523 Раздел ІІІ, п.3.7 Порядка: «Планирование, проведение и отчетность всех фаз клин")
15
Приказ МЗ Украины 191 от Исследования биодоступности и биоэквивалентности Руководство :2005 CPMP/EWP/QWP/1401/98 Note for Guidance on the Investigation of Bioavailability and Bioequivalence, 2001, ЕМЕА-2001 CPMP/EWP/QWP/1401/98/Rev.1 Guideline on the Investigation of Bioequivalence, ЕМА-2010

16
Содержит рекомендации по структуре и организации исследований биодоступности и биоэквивалентности, информацию об этих исследованиях, которую следует включать в регистрационные досье на ЛС. !!! Не распространяется на биологические (в том числе иммунобиологические препараты).

17
Приказ МЗ Украины от Надлежащая лабораторная практика (GLP) Проведение биоаналитической части клинических исследований по оценке биоэквивалентности лекарственных средств осуществляется в сертифицированной лаборатории с соблюдением основных требований и принципов надлежащей лабораторной практики (GLP) Директива 2004/10/ЕС Европейского Парламента и Совета ЕС
 Проведение биоаналитической части клинических исследований по оценке биоэквивалентности лекарственных средств осуществляется в сертифицированной лаборатории с соблюдением основ")
18
Рекомендации ВОЗ «Регуляторное руководство по взаимозаменяемости многоисточниковых (генерических) лекарственных средств» («Multisource (generic) pharmaceutical products guidelines on registration reguirements to establish interchangeability») WHO Expert Committee on Specifications for Pharmaceutical Preparations/WHO Technical Report Series, No. 937, 2006 Приказ МЗ Украины 663 от Перечень референтных лекарственных средств, рекомендуемых для применения при доказательстве эквивалентности (взаимозаменяемости) лекарственных средств
 лекарственных средств» («Multisource (generic) pharmaceutical products guidelines on registration reguirements to establish interchangeability») WHO Exp")
19
Выбор референтного ЛС для применения при доказательстве эквивалентности ЛС Утвержден Перечень референтных ЛС В случае отсутствия в утвержденном Перечне соответствующего референтного ЛС: "Руководство по выбору препаратов сравнения для оценки эквивалентности взаимозаменяемых многоисточниковых (генерических) ЛС" / WHO Expert Committee on Specifications for Pharmaceutical Preparations / WHO Technical Report Series, No. 902, 2002

20
РЕГЛАМЕНТИРУЕТ ТАКЖЕ: Раздел XІX – Критерии проведения дополнительных исследований ЛС при проведении экспертизы материалов регистрационного досье (в том числе, исследований биодоступности и эквивалентности генериков) Раздел XX – Порядок проведения дополнительных исследований ЛС (в том числе, исследований биодоступности и эквивалентности генериков) Рекомендации ВОЗ,Технический отчет ВОЗ, серия 937 (WHO Technical Report Series 937, Annex 7 Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchagebility). Приказ МЗ Украины 426 от г. в редакции приказа МЗ Украины 3 от г. (разделы XІX и XX) Порядок проведения экспертизы материалов на лекарственные средства, которые подаются на государственную регистрацию (перерегистрацию), а также экспертизы материалов о внесении изменений в регистрационные материалы на протяжении действия регистрационного свидетельства
 Раздел XX – Порядок проведения до")
21
Раздел XІX «Критерии проведения дополнительных исследований ЛС при проведении экспертизы материалов регистрационного досье» Пункт 4: «Дополнительные исследования эффективности и/или безопасности ЛС при проведении экспертизы материалов РД на ЛС проводятся в случае, если за результатами экспертизы отчетов проведенного (-ых) КИ определено: …проведенные КИ неадекватны для Подтверждения эффективности и безопасности ЛС…»; Пункт 6: «Дополнительные исследования биодоступности и эквивалентности ЛС при проведении экспертизы материалов РД на ЛС проводятся, если: - Данные представлены не в полном объеме для подтверждения эквивалентности ЛС… - Существует риск того, что возможные отличия биодоступности могут привести -к терапевтической неэквивалентности…» Приказ МЗ Украины 426 от г. в редакции приказа МЗ Украины 3 от г. (разделы XІX и XX) Порядок проведения экспертизы материалов на лекарственные средства, которые подаются на государственную регистрацию (перерегистрацию), а также экспертизы материалов о внесении изменений в регистрационные материалы на протяжении действия регистрационного свидетельства

22
Раздел XX «Порядок проведения дополнительных исследований ЛС» Пункт 4: «Дополнительные исследования биодоступности и эквивалентности генерических ЛС при проведении экспертизы материалов РД проводятся в соответствии с требованиями Порядка проведения КИ ЛС и экспертизы материалов КИ (приказ МЗ Украины от в редакции приказа МЗ Украины от )…» Пп – Определяет методы доказательства эквивалентности Пп – Определяет случаи отсутствия необходимости проведения исследований эквивалентности in vivo Пп – Определяет случаи необходимости проведения исследований эквивалентности in vivo Пп – Определяет условия проведения процедуры биовейвер … Приказ МЗ Украины 426 от г. в редакции приказа МЗ Украины 3 от г. (разделы XІX и XX) Порядок проведения экспертизы материалов на лекарственные средства, которые подаются на государственную регистрацию (перерегистрацию), а также экспертизы материалов о внесении изменений в регистрационные материалы на протяжении действия регистрационного свидетельства

23
«Исследования биодоступности и биоэквивалентности генерических лекарственных средств»

24
Методы доказательства эквивалентности генерических лекарственных средств

25
Отсутствие необходимости проведения исследований по оценке биоэквивалентности (ЛС с доказанной фармацевтической эквивалентностью) ЛС, которые фармацевтически эквивалентны и являются: - лекарственными формами для парентерального применения (внутривенного, подкожного, внутримышечного) в виде водных растворов; - растворами для орального применения (сиропы, эликсиры, настойки); - ушными или глазными ЛС в виде водных растворов; - препаратами местного действия в виде водных растворов; - ЛС системного действия в виде водных растворов для ректального или вагинального применения - водными растворами в форме ингаляционно- распылительных небулайзером препаратов, или назальные спреи, применяемые с помощью практически одинаковых устройств; - в форме порошков для приготовления растворов; - газами. ЛС, которые соответствуют этим критериям имеют 100 % биодоступность после введения - такое же действующее вещество(а) - в такой же молярной концентрации(ях) - такие же или подобные вспомогательные вещества в сравнимых с референтным препаратом концентрациях - такая же лек. форма - такой же путь введения
 ЛС, которые фармацевтически эквивалентны и являются: - лекарственными формами для парентерального применения (внутривенн")
26
Методы доказательства эквивалентности генерических ЛС: 1)Исследования in vivo (клинические исследования): -сравнительные фармакокинетические исследования (измеряется концентрация действующего вещества и / или его метаболита (ов) в доступной биологической жидкости, а именно: крови, плазме, сыворотке или моче для получения фармакокинетических показателей типа AUC и C мах, отражающих системное действие); -сравнительные фармакодинамические исследования с участием человека; -сравнительные клинические исследования. 2) Сравнительные исследования in vitro (БСК).
Исследования in vivo (клинические исследования): -сравнительные фармакокинетические исследования (измеряется концентрация действующего вещества и / или его метаболита (ов) в доступной биологиче")
27
Доказательство эквивалентности генерических ЛС методом сравнительных исследований in vitro Могут быть проведены для подтверждения эквивалентности лекарственных средств в твердых дозированных формах системного действия с немедленным высвобождением для орального применения Биовейвер - процедура проведения сравнительных исследований in vitro на основании биофармацевтической системы классификации с использованием теста "Растворение" для подтверждения эквивалентности генерического и референтного ЛС системного действия в твердой дозированной форме для орального применения с немедленным высвобождением с целью реестрации генерического ЛС без проведения исследований in vivo; Биофармацевтическая система классификации (БСК) - это научная система классификации действующих веществ на основе их растворимости в водных растворах и степени проникновения

28
СУБСТАНЦИЯ РАСТВОРИМОСТЬ ПРОНИЦАЕМОСТЬ ВЫСОКАЯ НИЗКАЯ ВЫСОКАЯ НИЗКАЯ ЛЕКАРСТВЕННАЯ ФОРМА РАСТВОРЕНИЕ ОЧЕНЬ БЫСТРОЕ БЫСТРОЕ МЕДЛЕННОЕ ПОКАЗАТЕЛИ, ИСПОЛЬЗУЕМЫЕ В БСК

29
БИОФАРМАЦЕВТИЧЕСКАЯ СИСТЕМА КЛАССИФИКАЦИИ (БСК) Отнесение лекарственной субстанции к тому или другому классу БКС позволяет решить вопрос о необходимости проведения исследований биоэквивалентности и биодоступности in vivo. Класс1: высокая растворимость, высокая степень проникновения Класс 2: низкая растворимость, высокая степень проникновения Класс 3: высокая растворимость, низкая степень проникновения Класс 4: низкая растворимость, низкая степень проникновения
 Отнесение лекарственной субстанции к тому или другому классу БКС позволяет решить вопрос о необходимости проведения исследований биоэквивалентности и биодоступности in vivo. Класс1: высокая растворимост")
30
Исследования in vivo нужно проводить в случае, когда существует риск того, что возможные отличия в биодоступности могут привести к терапевтической неэквивалентности препарата Факторы, влияющие на биодоступность: Физико-химические свойства действующего вещества (степень дисперсности, полиморфизм) Вспомогательные вещества (состав покрытия таблеток и капсул) Технологический процесс (грануляция порошков) Условия хранения и тип упаковки (стекло, пластмасса, бумага и др.) Исследования in vivo

31
Необходимость проведения исследований эквивалентности in vivo ( клинических исследований по оценке биоэквивалентности) а) оральные ЛС системного действия с немедленным высвобождением, когда к ним применяется хотя бы один из критериев: применения ЛС в неотложных состояниях узкое терапевтическое окно (границы эффективности / безопасности) крутая кривой доза-ответ документально подтвержденные проблемы по биодоступности или бионеэквивалентности, которые связаны с действующим в-вом, или его лекарственными формами данные о том, что на биоэквивалентность может влиять полиморфизм действующего в-ва, вспомогательные в-ва и/или технологические процессы, используемые в производстве
 а) оральные ЛС системного действия с немедленным высвобождением, когда к ним применяется хотя бы один из критериев: применения ЛС в")
32
Необходимость проведения исследований эквивалентности in vivo (клинических исследований по оценке биоэквивалентности) б) неоральные и непарентеральные препараты системного действия (трансдермальные пластыри, суппозитории, никотиновые жевательные резинки и т.д.); в) препараты с модифицированным высвобождением системного действия; г) препараты с фиксированной комбинацией системного действия, в которых, как минимум, для одного действующего в-ва требуется проведение исследования in vivo; д) препараты не в виде растворов несистемного действия (для орального, назального, офтальмологического, дерматологического, ректального или вагинального применения) е) ЛС, которые имеют отличия от референтного: эфир, сложный эфир, изомер, рацемат, комплексы или продукты дериватизации действующего вещества, т.к. эти отличия могут обусловить разную биодоступность.
 б) неоральные и непарентеральные препараты системного действия (трансдермальные пластыри, суппозитории, никотиновые жевательные рези")
33
КИ по оценке биоэквивалентности ЛС В классическом случае: КИ для оценки биоэквивалентности - это исследование биодоступности с использованием фармакокинетических конечных точек – сравнительное фармакокинетическое исследование; по сути - сравнительное изучение биодоступности. Проводится если возможно получить измеряемые концентрации действующего вещества в биологической жидкости, такой как плазма Если это невозможно, то: - КИ с использованием фармакодинамических конечных точек; - КИ с использованием других клинических конечных точек;

34
Сравнительные фармакокинетические исследования по оценке биоэквивалентности Отчет исследования по оценке биоэквивалентности генерического ЛС Статистический анализ GSP Клинические исследования на здоровых добровольцах GCP Биоаналитическая оценка GLP

35
Особенности сравнительных фармакокинетических исследований по оценке биоэквивалентности ЛС –Нетерапевтические клинические исследования –Субъекты исследования – здоровые добровольцы, но: "Если известно, что исследуемое действующее вещество имеет побочные эффекты, а фармакологическое действие или риск считаются неприемлемыми для здоровых добровольцев, может быть необходимо проведение таких исследований с участием больных при соблюдении соответствующих мер и под наблюдением" (приказ МЗ Украины 191 от г., п ) Например, исследуемое ЛС предназначено для использования в онкологии, для лечения ВИЧ-инфицированных пациентов, или является психотропным ЛС –Объект исследования – генерическое лекарственное средство

36
Количество добровольцев Возраст ЕС (EMA)Не менее 12>18 ВОЗНе менее УкраинаНе менее Особенности сравнительных фармакокинетических исследований по оценке биоэквивалентности ЛС
Не менее 12>18 ВОЗНе менее 1218 - 55 УкраинаНе менее 1218 - 55 Особенности сравнительных фармакокинетических исследований по оценке биоэквивалентности ЛС")
37
Спавнение требований к стандартизации условий пребывания добровольцев в клинике ЕС, EMA Состав и время приема пищи Употребление жидкости при приеме препарата (150 мл) Отдельные палаты Курение Блок интенсивной терапии Физическая активность Добровольное желание ВОЗ Употребление жидкости при приеме препарата ( мл) УкраинаНе отличаются
 Отдельные палаты Курение Блок интенсивной терапии Физическая активность Добровольно")
38
38 Особенности диеты для здоровых добровольцев в исследованиях биоэквивалентности период голодания водный режим стандартные ограничения (кофеин, алкоголь, цитрусовые …) ограничения, предусмотренные протоколом (низкое содержание белка, животных жиров …) унификация диеты при исследовании на нескольких клинических базах Особенности диеты для здоровых добровольцев в исследованиях биоэквивалентности период голодания водный режим стандартные ограничения (кофеин, алкоголь, цитрусовые …) ограничения, предусмотренные протоколом (низкое содержание белка, животных жиров …) унификация диеты при исследовании на нескольких клинических базах Особенности сравнительных фармакокинетических исследований по оценке биоэквивалентности ЛС
 ограничения, предусмотренные протоколом (низкое содержание белка, животных жиров")
39
Клинические исследования по оценке биоэквивалентности, как и другие КИ ЛС, регулируются действующим Порядком проведения клинических исследований лекарственных средств и экспертизы материалов клинических исследований (приказ МЗ Украины от с изменениями, внесенными приказом МЗ Украины от ) КИ может начинаться при наличии положительного заключения Центра, утвержденного МЗ, протокола Комиссии по вопросам этики при ЛПУ, где проводятся КИ и при условии оформления договорных отношений между всеми юридическими и физическими лицами, которые вовлечены в проведение КИ, согласно действующему законодательству" (Раздел IX, пункт 9.1 Порядка) Для получения заключения Центра, утвержденного МЗ относительно проведения КИ заявитель подает в ГЭЦ пакет документов в соответствии с разделом VII Порядка

40
Исследуемое ЛС - лекарственная форма активной субстанции или плацебо, которая изучается или используется для сравнения в КИ, включая препараты, на которые уже выдано регистрационное удостоверение, но они используются или производятся (составленные или упакованные) другим способом по сравнению с зарегистрированной лекарственной формой, или используются для получения дополнительной информации о зарегистрированной форме ЛС (Раздел ІІ Порядка, приказ от с изменениями, внесенными приказом МЗ от ) В КИ референтное ЛС – исследуемое ЛС! На исследуемое ЛС предоставляется подтверждение, что работы на производственном участке или объекте, который занят в производстве данного ЛС, проводятся с соблюдением принципов надлежащей производственной практики с предоставлением сертификата GMP или письменного официального заявления Уполномоченного лица по качеству (производителя) или копию документа, выданного Государственной службой лекарственных средств Украины (раздел VII, пункт Порядка)

41
Требования к ЛПУ / месту проведения клинического исследования Клинические исследования проводятся в ЛПУ: в случае если *: при ЛПУ создана и действует комиссия по вопросам этики при ЛПУ; имеется необходимая база для оказания квалифицированной неотложной медицинской помощи испытуемым в случае возникновения осложнений во время проведения КИ и действующий блок интенсивной терапии или реанимационное отделение в зависимости от особенностей КИ; в подразделениях ЛПУ (лаборатория, отделение функциональной диагностики) проводится метрологический контроль, в порядке, предусмотренном законодательством; есть условия для хранения исследуемых ЛС (согласно условиям хранения, указанных при маркировке лекарственных средств или в протоколе КИ) и документации, которая относится к КИ; есть возможность включать необходимое количество испытуемых, в соответствии с протоколом; условия хранения материалов КИ (в архивном помещении) не менее 15 лет после завершения КИ * - раздел V Порядка приказ МЗ от р. 690 с изменениями, внесенными приказом МЗ от

42
Продолжение Кроме этого, для КИ по оценке биоэквивалентности должны выполняться следующие требования : имеются отдельные палаты для испытуемых и условия, необходимые для круглосуточного наблюдения за их состоянием; отдельная манипуляционная, столовая и санитарная комната для испытуемых; условия хранения биологических образцов при проведении клинических исследований.

43
Биоаналитическая часть КИ по оценке биоэквивалентности ЛС проводится в соответствующей лаборатории, имеющей сертификат аккредитации и отвечающей требованиям Руководства "Лекарственные средства. Надлежащая лабораторная практика В такой лаборатории: имеется необходимое оборудование для проведения фармакокинетических исследований; созданы надлежащие условия для хранения образцов биопроб; ведется вся необходимая документация; исследования осуществляются в соответствии со стандартными операционными процедурами.

44
44 Хранение биоматериала холодильник криопробирки криобоксы криолейблы криомаркер Хранение биоматериала холодильник криопробирки криобоксы криолейблы криомаркер

45
Требования к исследователям * Исследователи, которые будут принимать участие в клиническом исследовании, должны: иметь достаточную профессиональную подготовку; знать международные требования в сфере проведения клинических исследований, в частности надлежащую клиническую практику (GCP) и нормативно-правовые акты о проведении клинических исследований в Украине; работать в ЛПУ, где планируется проведение клинического исследования (в случае если исследователь является сотрудником кафедры высшего медицинского учебного заведения, необходимо наличие договора о сотрудничестве между высшим медицинским учебным заведением и ЛПУ); исследователи, которые привлекаются к проведению клинических исследований I фазы и биоэквивалентности лекарственных средств, должны иметь опыт проведения клинических исследований, что подтверждается информацией, приведенной в автобиографиях (curriculum vitae) * - раздел V Порядка, приказ МЗ от р. 690 c изменениями, внесенными приказом МЗ от

46
Заключительный отчет о проведенном клиническом исследовании по оценке биоэквивалентности Результаты клинических исследований по оценке биоэквивалентности лекарственных средств в виде отчетов предоставляются в составе регистрационного досье: - Часть IV "Клиническая документация" - Модуль 5 "Отчет о КИ" (Формат общетехнического документа) СРMP/ICH/137/95 (Е3) Руководящие указания по структуре и содержанию отчетов по клиническому исследованию» Приложение 13 Порядка проведения КИ ЛС и экспертизы материалов КИ (утвержден приказом МЗ Украины От в редакции приказа МЗ Украины от )

47
Структура регистрационного досье, в том числе, части 4 «клиническая документация», модуля 5 «отчеты о КИ» Требования к материалам досье, в том числе к части 4, модулю 5 (пункты 5.3.3, 5.3.4, 5.3.5: отчеты о фармакокинетических, фармакодинамических исследованиях у человека, об исследовании эффективности и безопасности) Заключительный отчет о проведенном клиническом исследовании по оценке биоэквивалентности Приказ МЗ Украины 426 от г. в редакции приказа МЗ Украины 3 от г. Порядок проведения экспертизы материалов на лекарственные средства, которые подаются на государственную регистрацию (перерегистрацию), а также экспертизы материалов о внесении изменений в регистрационные материалы на протяжении действия регистрационного свидетельства

48
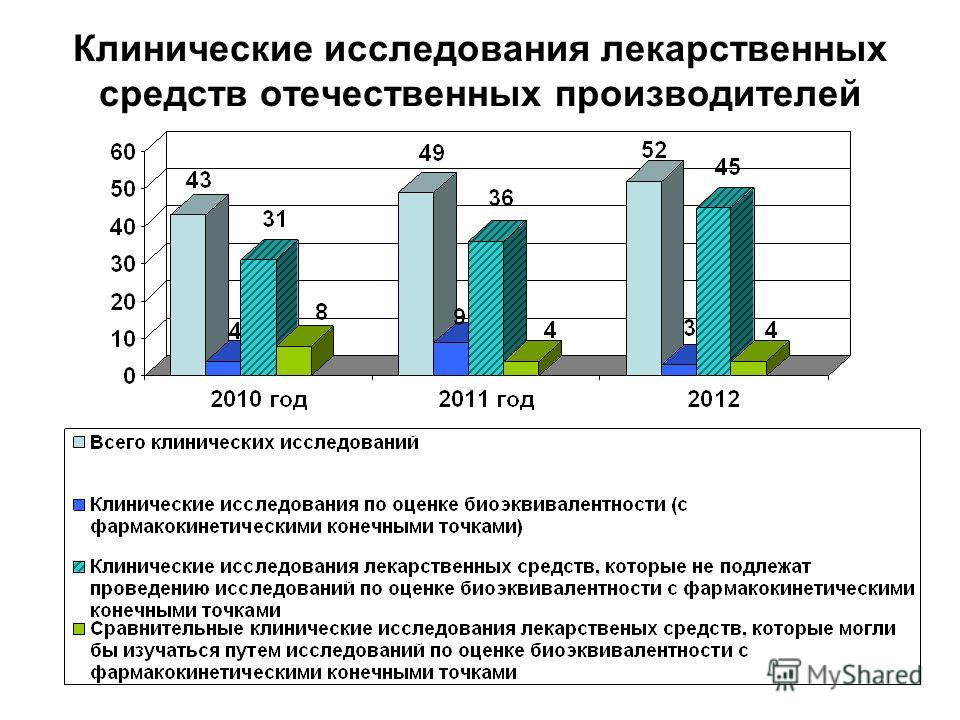
Клинические исследования лекарственных средств отечественных производителей

50
СПАСИБО ЗА ВНИМАНИЕ!

Еще похожие презентации в нашем архиве:














 клинических испытаний в Украине Распутняк Сергей Сергеевич Заместитель директора Департамента доклинических и клинических.")





