Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
MELAS-СИНДРОМ КУЗЕНКОВА Л.М., ГЛОБА О.В. Отделение психоневрологии НИИ педиатрии, Научный Центр Здоровья Детей РАМН, Москва

2
Митохондриальные нарушения – это обширная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий, нарушением тканевого дыхания и, как следствие, недостаточностью энергетического обмена. Митохондриальные нарушения – это обширная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий, нарушением тканевого дыхания и, как следствие, недостаточностью энергетического обмена. Нарушения клеточной энергетики приводят к полисистемным заболеваниям. В первую очередь страдают органы и ткани, наиболее энергозависимые – нервная система (энцефалопатии, полиневропатии), мышечная система (миопатии), сердце (кардиомиопатии), почки, печень, эндокринная система и другие. До самого недавнего времени все эти заболевания определялись под многочисленными масками других патологических форм. Нарушения клеточной энергетики приводят к полисистемным заболеваниям. В первую очередь страдают органы и ткани, наиболее энергозависимые – нервная система (энцефалопатии, полиневропатии), мышечная система (миопатии), сердце (кардиомиопатии), почки, печень, эндокринная система и другие. До самого недавнего времени все эти заболевания определялись под многочисленными масками других патологических форм. К настоящему времени выявлено более 200 заболеваний, причиной которых являются мутации митохондриальной ДНК. Митохондриальные болезни могут быть обусловлены как патологией митохондриального, так и ядерного генома. В связи с углублением знаний молекулярной генетики выявляются все новые и новые мутации. К настоящему времени выявлено более 200 заболеваний, причиной которых являются мутации митохондриальной ДНК. Митохондриальные болезни могут быть обусловлены как патологией митохондриального, так и ядерного генома. В связи с углублением знаний молекулярной генетики выявляются все новые и новые мутации.

4
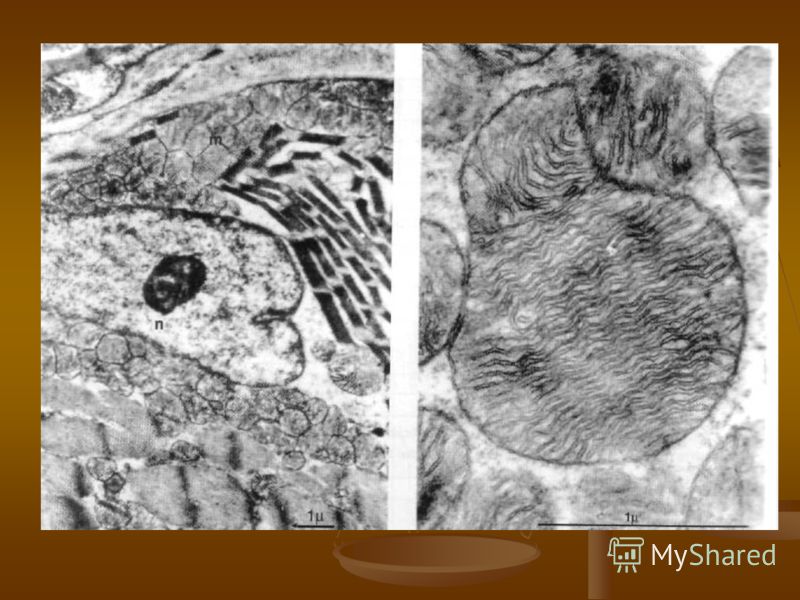
В клетке находятся от нескольких сотен до тысяч органелл – митохондрий, содержащих от 2 до 10 кольцевых молекул митохондриальной ДНК, способных к репликации, транскрипции и трансляции, независимо от ядерной ДНК.

6
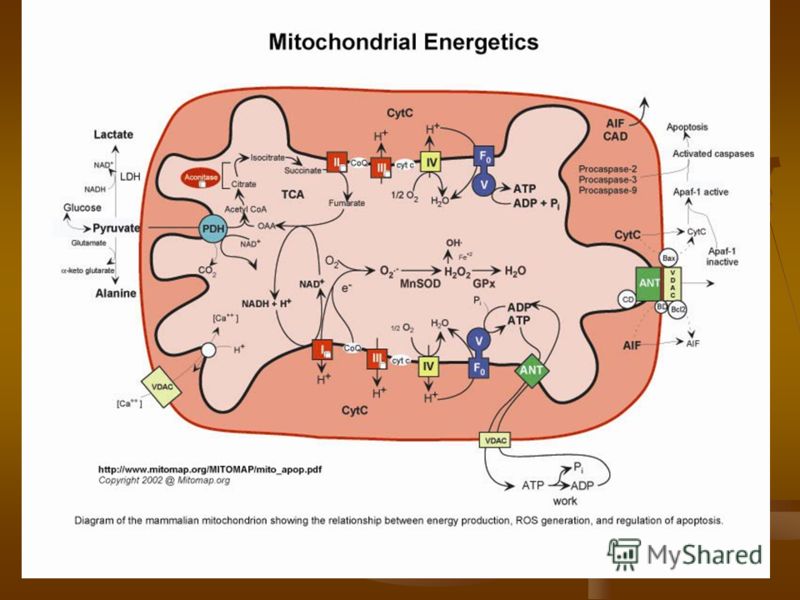
Дыхательная цепь локализуется на внутренней мембране митохондрий и включает в себя пять мультиферментных комплексов, каждый из которых состоит из нескольких десятков субъединиц. Конечным результатом окислительного фосфорилирования, происходящего в комплексах 1-Y является производство энергии (АТФ). Дыхательная цепь локализуется на внутренней мембране митохондрий и включает в себя пять мультиферментных комплексов, каждый из которых состоит из нескольких десятков субъединиц. Конечным результатом окислительного фосфорилирования, происходящего в комплексах 1-Y является производство энергии (АТФ).

7
ТИП НАСЛЕДОВАНИЯ Митохондриальная генетика отличается от менделевской в трех важнейших аспектах: Митохондриальная генетика отличается от менделевской в трех важнейших аспектах: Материнское наследование - всю цитоплазму, вместе с находящимися в ней органеллами потомки получают вместе с яйцеклеткой. Гетероплазмия- одновременное существование в клетке нормального (дикий) и мутантного типов ДНК Митотическая сегрегация- оба типа мтДНК в процессе деления клетки могут распределяться случайным образом между дочерними клетками.

8
Заболевания, обусловленные мутациями митохондриальной ДНК, характеризуются выраженным полиморфизмом клинических симптомов и вариабельностью течения. В этой связи принципиально важное значение для клинической практики имеет определение критериев диагноза данной группы болезней с целью своевременного назначения адекватной терапии и потенциального снижения инвалидизации больных. Заболевания, обусловленные мутациями митохондриальной ДНК, характеризуются выраженным полиморфизмом клинических симптомов и вариабельностью течения. В этой связи принципиально важное значение для клинической практики имеет определение критериев диагноза данной группы болезней с целью своевременного назначения адекватной терапии и потенциального снижения инвалидизации больных.

9
Ведущими неврологическими синдромами при заболеваниях, обусловленных мутациями митохондриальной ДНК, являются: миопатический симптомокомплекс (непереносимость физических нагрузок, мышечная слабость, снижение мышечного тонуса), судороги, мозжечковый синдром (атаксия, интенционный тремор), поражение глазодвигательных нервов (птоз, наружная офтальмоплегия), полиневропатия. Ведущими неврологическими синдромами при заболеваниях, обусловленных мутациями митохондриальной ДНК, являются: миопатический симптомокомплекс (непереносимость физических нагрузок, мышечная слабость, снижение мышечного тонуса), судороги, мозжечковый синдром (атаксия, интенционный тремор), поражение глазодвигательных нервов (птоз, наружная офтальмоплегия), полиневропатия. О наличии у ребенка митохондриальной патологии могут свидетельствовать миоклонические или мультифокальные судороги, резистентные к антиконвульсантам, черепно-лицевая дисморфия, дисметаболические проявления (рвота, эпизоды летаргии, комы), дыхательные нарушения (апноэ, гипервентиляция, тахипноэ), вовлечение в патологический процесс ЦНС, мышц, сердца,печени,почек, а также прогрессирующее течение заболевания. О наличии у ребенка митохондриальной патологии могут свидетельствовать миоклонические или мультифокальные судороги, резистентные к антиконвульсантам, черепно-лицевая дисморфия, дисметаболические проявления (рвота, эпизоды летаргии, комы), дыхательные нарушения (апноэ, гипервентиляция, тахипноэ), вовлечение в патологический процесс ЦНС, мышц, сердца,печени,почек, а также прогрессирующее течение заболевания.
, судороги, мозжечковый с")
10
КРИТЕРИИ ДИАГНОСТИКИ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Абсолютным критерием диагноза митохондриальной болезни являются результаты молекулярно-генетического исследования Mt-ДНК с выявлением конкретных мутаций. Однако более доступными являются биохимические тесты (лакта-пируват-ацидоз), а также биопсия скелетных мышц с проведением специфических гистохимических реакций, выявление феномена рваных красных волокон. Абсолютным критерием диагноза митохондриальной болезни являются результаты молекулярно-генетического исследования Mt-ДНК с выявлением конкретных мутаций. Однако более доступными являются биохимические тесты (лакта-пируват-ацидоз), а также биопсия скелетных мышц с проведением специфических гистохимических реакций, выявление феномена рваных красных волокон. Истинная классификация митохондриальных болезней основана на молекулярно-генетических данных, однако на практике применяется клинико-синдромологический подход. Трудности клинической диагностики связаны с мультисистемным поражением и выраженным фенотипическим полиморфизмом. Истинная классификация митохондриальных болезней основана на молекулярно-генетических данных, однако на практике применяется клинико-синдромологический подход. Трудности клинической диагностики связаны с мультисистемным поражением и выраженным фенотипическим полиморфизмом.

11
НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЮЩИЕСЯ В ДЕТСКОМ ВОЗРАСТЕ МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ Синдром MELAS – митохондриальная энцефаломитопатия, лактат-ацидоз и инсультоподобные пароксизмы Синдром MELAS – митохондриальная энцефаломитопатия, лактат-ацидоз и инсультоподобные пароксизмы Синдром MERRF – миоклонус-эпилепсия, рваные красные волокна Синдром MERRF – миоклонус-эпилепсия, рваные красные волокна KSS – синдром Кернса-Сейра – характеризуется птозом, офтальмоплегией, пигментным ретинитом, атаксией, нарушением сердечного проведения KSS – синдром Кернса-Сейра – характеризуется птозом, офтальмоплегией, пигментным ретинитом, атаксией, нарушением сердечного проведения Синдром NARP – нейропатия, атаксия, пигментный ретинит Синдром NARP – нейропатия, атаксия, пигментный ретинит Синдром Лея – подострая некротизирующая энцефаломиелопатия Синдром Лея – подострая некротизирующая энцефаломиелопатия Синдром Альперса – прогрессирующее нарушение ментальных функций, гипотония, судороги, нарушение зрения и слуха Синдром Альперса – прогрессирующее нарушение ментальных функций, гипотония, судороги, нарушение зрения и слуха

12
МИТОХОНДРИАЛЬНАЯ ЭНЦЕФАЛОМИОПАТИЯ, ЛАКТАТ-АЦИДОЗ И ИНСУЛЬТОПОДОБНЫЕ ПАРОКСИЗМЫ (MELAS) Мультисистемное заболевание, описанное в 1984 г Pavlakis SG et all, характеризующееся - Инсультоподобными пароксизмами, возникающими в молодом возрасте (до 40 лет) - Энцефалопатией, характеризующейся судорогами, деменцией - Митохондриальной миопатией с лактат-ацидозом,рваными красными волокнами Дополнительные проявления: низкий рост, кардиомиопатия, кальцинаты базальных ганглиев, миоклонус, атаксия, мигренеподобные головные боли, атрофия зрительных нервов, пигментная ретинопатия, потеря слуха, офтальмоплегия, диабет, нарушение сердечной проводимости, гастроинтестинальные нарушения, нефропатия, тошнота Дополнительные проявления: низкий рост, кардиомиопатия, кальцинаты базальных ганглиев, миоклонус, атаксия, мигренеподобные головные боли, атрофия зрительных нервов, пигментная ретинопатия, потеря слуха, офтальмоплегия, диабет, нарушение сердечной проводимости, гастроинтестинальные нарушения, нефропатия, тошнота Основная характеристика этого заболевания – инсультоподобные пароксизмы, наиболее часто локализующиеся в затылочной области, приводящие к гемианопсии или корковой слепоте. Однако, зоны могут быть различными.
 Мультисистемное заболевание, описанное в 1984 г Pavlakis SG et all, характеризующееся - Инсультоподобными пароксизмами, возникающими в молодом возрасте (до 40 лет)")
14
ГЕНЕТИКА СИНДРОМА Около 15 мтДНК мутаций ассоциировано с синдромом MELAS. Примерно 80% из них имеют мутацию A3243G tRNA leu. Однако,корреляция между генотипом и фенотипом не является прямой. Может присутствовать как весь симптомокомплекс, так и отдельные проявления. Около 15 мтДНК мутаций ассоциировано с синдромом MELAS. Примерно 80% из них имеют мутацию A3243G tRNA leu. Однако,корреляция между генотипом и фенотипом не является прямой. Может присутствовать как весь симптомокомплекс, так и отдельные проявления. Большинство мутаций находится в tRNA генах, однако они также могут находиться в полипептид-кодирующих генах, включая субъединицу Ш cytochrom c oxidase (COX) и субъединицы 1,5 и 6 1 комплекса. Большинство мутаций находится в tRNA генах, однако они также могут находиться в полипептид-кодирующих генах, включая субъединицу Ш cytochrom c oxidase (COX) и субъединицы 1,5 и 6 1 комплекса. Важно помнить, что G13513A мутация в субъединицу 5 комплекса 1 (ND5) - частая причина клинических проявлений MELAS, наследственной нейропатии Лебера и синдрома Лея (Menkes JH.et.al,2000, DiMauro S et al, 2006). Важно помнить, что G13513A мутация в субъединицу 5 комплекса 1 (ND5) - частая причина клинических проявлений MELAS, наследственной нейропатии Лебера и синдрома Лея (Menkes JH.et.al,2000, DiMauro S et al, 2006). Встречаются случаи, когда первыми или единственными проявлениями митохондриальной болезни был инсульт, при этом при ДНК диагностике выявлялись мутации A3243G, A8344G (характерные для MERRF) и мутации, ассоциированные с оптической нейропатией Лебера (Martinez-Fernandez E. et al,2001) Встречаются случаи, когда первыми или единственными проявлениями митохондриальной болезни был инсульт, при этом при ДНК диагностике выявлялись мутации A3243G, A8344G (характерные для MERRF) и мутации, ассоциированные с оптической нейропатией Лебера (Martinez-Fernandez E. et al,2001)

15
ОСОБЕННОСТИ ИНСУЛЬТА ПРИ СИНДРОМЕ MELAS Частая локализация в затылочной области, приводящая к гемианопсии или корковой слепоте. Частая локализация в затылочной области, приводящая к гемианопсии или корковой слепоте. Инсультоподобные пароксизмы атипичные, т.е. поражают в основном молодых, часто провоцируются заболеваниями, сопровождающимися фебрильной температурой, мигренеподобной головной болью, возникают после судорог Инсультоподобные пароксизмы атипичные, т.е. поражают в основном молодых, часто провоцируются заболеваниями, сопровождающимися фебрильной температурой, мигренеподобной головной болью, возникают после судорог Очаги часто лежат вне региона крупных церебральных артерий, чаще поражая кору или глубинные структуры белого вещества головного мозга (Mathews PM et al 1991) Очаги часто лежат вне региона крупных церебральных артерий, чаще поражая кору или глубинные структуры белого вещества головного мозга (Mathews PM et al 1991) Острые MELAS очаги могут флюктуировать, мигрировать или даже исчезать. Кальцификаты базальных ганглиев – частая находка (Sue SM et al, 1998) Острые MELAS очаги могут флюктуировать, мигрировать или даже исчезать. Кальцификаты базальных ганглиев – частая находка (Sue SM et al, 1998) Ангиография подтверждает отсутствие патологии со стороны крупных сосудов (Hirano M,Pavlakis S 1994). Ангиография подтверждает отсутствие патологии со стороны крупных сосудов (Hirano M,Pavlakis S 1994).

16
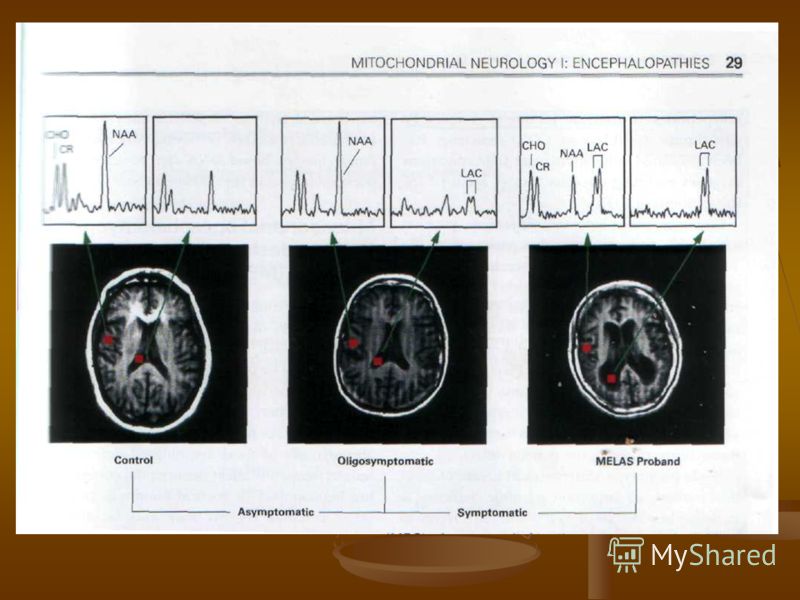
Количественный анализ церебрального кровотока, исследованный с помощью ксенон КТ выявил нормальный или усиленный кровоток в зоне острого повреждения и непораженных областях, что предполагает, что инсультоподобные проявления возникают не из-за фокальной ишемии (Morita K, et.al 1989, Ooiwa Y et al.1993) Количественный анализ церебрального кровотока, исследованный с помощью ксенон КТ выявил нормальный или усиленный кровоток в зоне острого повреждения и непораженных областях, что предполагает, что инсультоподобные проявления возникают не из-за фокальной ишемии (Morita K, et.al 1989, Ooiwa Y et al.1993) SPECT обычно выявляет снижение аккумуляции вводимого вещества в зонах повреждения, возможно, в связи с потерей метаболически активных клеток (Saton M et al 1991, Watanabe Y et al 1998) SPECT обычно выявляет снижение аккумуляции вводимого вещества в зонах повреждения, возможно, в связи с потерей метаболически активных клеток (Saton M et al 1991, Watanabe Y et al 1998) Имеются интересные наблюдения об увеличении поглощения вещества при SPECT и увеличение регионального кровотока при PET исследованиях за 3-6 дней до появления инсульта (Takahashi S et al 1998). Возможно, что повышение кровотока до острого инсультоподобного эпизода может отражать локализованный ответ на повышение уровня лактата, фокальное повышение метаболизма или повреждение вазорегуляции. Имеются интересные наблюдения об увеличении поглощения вещества при SPECT и увеличение регионального кровотока при PET исследованиях за 3-6 дней до появления инсульта (Takahashi S et al 1998). Возможно, что повышение кровотока до острого инсультоподобного эпизода может отражать локализованный ответ на повышение уровня лактата, фокальное повышение метаболизма или повреждение вазорегуляции. MRS выявляет повышение лактата в головном мозга, отражающее компенсаторное повышение анаэробного гликолиза. Выялено также, что содержание вентрикулярного лактата находится в прямой корреляционной зависимости от неврологическогодефицита при MELAS (Dubeau F et al 2000, Kaufmann P et al 2004) MRS выявляет повышение лактата в головном мозга, отражающее компенсаторное повышение анаэробного гликолиза. Выялено также, что содержание вентрикулярного лактата находится в прямой корреляционной зависимости от неврологическогодефицита при MELAS (Dubeau F et al 2000, Kaufmann P et al 2004)

18
Т.О. нейрорадиологические исследования при синдроме MELAS выявили, что атипичные инсультоподобные пароксизмы возникают не столько от острой ишемии, сколько от метаболической дисфункции со снижением окислительного фосфорилирования, повышением кровня лактата и снижением синтеза АТФ ( DiMauro S. et al 2006). Т.О. нейрорадиологические исследования при синдроме MELAS выявили, что атипичные инсультоподобные пароксизмы возникают не столько от острой ишемии, сколько от метаболической дисфункции со снижением окислительного фосфорилирования, повышением кровня лактата и снижением синтеза АТФ ( DiMauro S. et al 2006).

19
GLUTAMATE Na+ GLUTAMATE Metabo Kainat AMPANMDA GLUTAMINE SYNTHASE GLIA Са 2+ GLYCINE POSTSYNAPTIC MEMBRANE GLUTAMINE glucosaepyruvate Krebs` cycle α ketoglutarate ATPADP

20
Наши генетические исследования

21
Генетические исследования Генетические исследования, проведенные у 24 детей, выявили следующее: A3243G, A11084G (MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis and stroke- like episodes syndrome), мутации в ATPsynthase 6, цитохром С оксидаза, 16S rRNA (T3197C), tRNA lysine, 2 ND A5069G/N235D, 1ND,множественные точечные мутации mtDNA. Генетические исследования, проведенные у 24 детей, выявили следующее: A3243G, A11084G (MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis and stroke- like episodes syndrome), мутации в ATPsynthase 6, цитохром С оксидаза, 16S rRNA (T3197C), tRNA lysine, 2 ND A5069G/N235D, 1ND,множественные точечные мутации mtDNA.
, мутации в ATPsynthase 6, цитохром С о")
22
16S rRNA мутации представлены гипогликемией, эпилептиформными ЭЭГ-изменениями, судорогами, инсультоподобными пароксизмами. 16S rRNA мутации представлены гипогликемией, эпилептиформными ЭЭГ-изменениями, судорогами, инсультоподобными пароксизмами. Первыми проявлениями мутации АТФ синтазы 6 была дилятационная кардиомиопатия. В дальнейшем возникали инсультоподобные пароксизмы и судороги. Первыми проявлениями мутации АТФ синтазы 6 была дилятационная кардиомиопатия. В дальнейшем возникали инсультоподобные пароксизмы и судороги.

23
Ребенок с перенесенным в возрасте 12 лет инсультом, с явлениями гемипареза, мышечной гипотонией, низким ростом, интеллектуальным дефицитом. Через 6 месяцев перенесла инсульт повторно. Ребенок с перенесенным в возрасте 12 лет инсультом, с явлениями гемипареза, мышечной гипотонией, низким ростом, интеллектуальным дефицитом. Через 6 месяцев перенесла инсульт повторно.

25
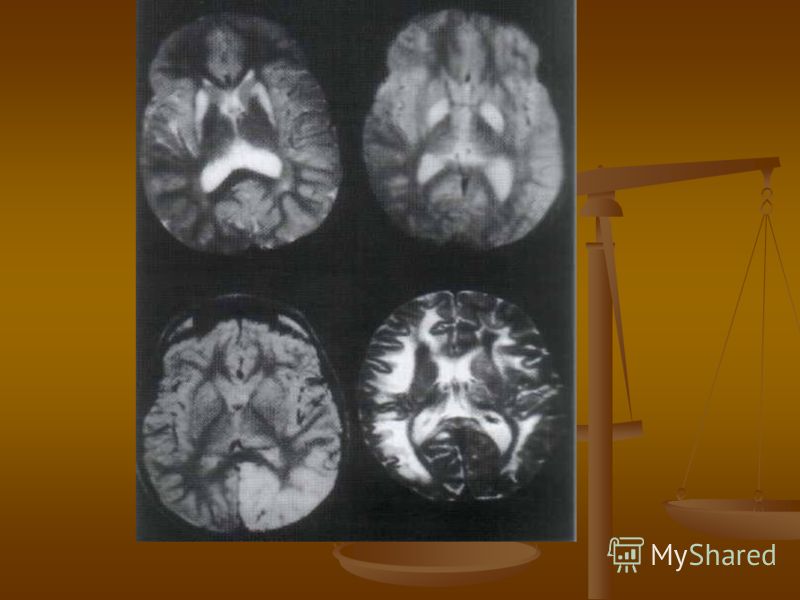
МРТ исследования выявило корковую субатрофию, перивентрикулярную лейкомаляцию, инсультоподобные изменения.

27
SPECT проведенный у 10 детей, выявил снижение и неравномерность перфузии мозговой ткани.

29
Ребенок с задержкой физического развития, мышечной гипотонией, коарктационным синдромом,ДКМП с выявленой мутацией A3243G

30
ЭЭГ 11-летней пациентки до метаболического лечения (ребенок получает депакин)
")
31
ЭЭГ той же пациентки через 4 месяца метаболической терапии ( в дополнении к антиконвульсантной).
.")
32
ЛЕЧЕНИЕ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Нет достоверных подтверждений, что какая-либо терапия может быть эффективна в отношении митохондриальных болезней. Различные антиоксиданты, витамины, кофакторы дыхательной цепи применяются у детей, страдающих обменными расстройствами. Нет достоверных подтверждений, что какая-либо терапия может быть эффективна в отношении митохондриальных болезней. Различные антиоксиданты, витамины, кофакторы дыхательной цепи применяются у детей, страдающих обменными расстройствами. При синдроме MELAS лечение должно быть направлено на лечение судорог, эндокринных расстройств, устранение последствий инсульта. При синдроме MELAS лечение должно быть направлено на лечение судорог, эндокринных расстройств, устранение последствий инсульта. Т.к. уровень лактата часто коррелирует с тяжестью неврологических проявлений, целесообразно применять дихлорацетат (у нас в стране используется демифосфон) для снижения уровня лактата (Kaufmann P et al, 2006) Т.к. уровень лактата часто коррелирует с тяжестью неврологических проявлений, целесообразно применять дихлорацетат (у нас в стране используется демифосфон) для снижения уровня лактата (Kaufmann P et al, 2006) В исследованиях японских авторов Koga Y et al, (2002, 2005,2008) использовался в/в L-аргинин, предшественник NO, для стимуляции вазодилятации в остром периоде инсульта с хорошими результатами, а также пероральное его применение для снижения тяжести последующих эпизодов В исследованиях японских авторов Koga Y et al, (2002, 2005,2008) использовался в/в L-аргинин, предшественник NO, для стимуляции вазодилятации в остром периоде инсульта с хорошими результатами, а также пероральное его применение для снижения тяжести последующих эпизодов

33
Лечение КоэнзимQ10 – 120 мг/сут КоэнзимQ10 – 120 мг/сут L-карнитин – от 10 мг до 1-2 г/сут L-карнитин – от 10 мг до 1-2 г/сут Вит В1 400 мг/сут Вит В1 400 мг/сут Вит В2 100 мг/сут Вит В2 100 мг/сут Вит С до 1 г/сут Вит С до 1 г/сут Вит Е 400 МЕ в сут Вит Е 400 МЕ в сут Янтарная кислота от 25 мг до 1.5 г в сутки Янтарная кислота от 25 мг до 1.5 г в сутки Демифосфон 15% 1 мл на 5 кг веса Демифосфон 15% 1 мл на 5 кг веса Реамберин в/в, цитофлавин в/в и перорально Реамберин в/в, цитофлавин в/в и перорально Кортикостероиды в/м Кортикостероиды в/м Антиконвульсанты (исключая вальпроевую кислоту и ее производные, ограничивая барбитураты) Антиконвульсанты (исключая вальпроевую кислоту и ее производные, ограничивая барбитураты)

34
Важность своевременной диагностики митохондриальных болезней, поиска клинических и параклинических критериев этих заболеваний на предварительном, догенетическом этапе необходимо для подбора адекватной метаболической терапии и предотвращения ухудшения состояния или инвалидизации больных с этими редкими заболеваниями. Важность своевременной диагностики митохондриальных болезней, поиска клинических и параклинических критериев этих заболеваний на предварительном, догенетическом этапе необходимо для подбора адекватной метаболической терапии и предотвращения ухудшения состояния или инвалидизации больных с этими редкими заболеваниями.

Еще похожие презентации в нашем архиве:



 описан Фукухама в 1980 г.")













. Мукополисахаридоз II типа (МПС II, OMIM - 309900), или синдром Хантера - это лизосомная болезнь накопления.")



