Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
Наследственные нейромышечные заболевания 586 ОМ

2
Нервно-мышечные заболевания характеризуются нарушением движений и прогрессирующей атрофией мышц. Выделяют: прогрессирующие мышечные дистрофии (миопатии); спинальные амиотрофии; наследственные полиневропатии; миотонии и миоплегии
; спинальные амиотрофии; наследственные полиневропатии; миотонии и миоплегии")
3
Наследственные болезни мышц, мышечные дистрофии и миопатии

4
Клиническая классификация прогрессирующих мышечных дистрофий (ПМД) основана на характере распространения мышечных атрофий и парезов – конечностно-поясные, лице-лопаточно-плечевые, дистальные, окулофаренгиальные.
 основана на характере распространения мышечных атрофий и парезов – конечностно-поясные, лице-лопаточно-плечевые, дистальные, окулофаренгиальные.")
5
Морфологически в мышечных волокнах при миопатиях выявляются дистрофические и некротические изменения, разрастание соединительной ткани, диффузная разнокалиберность миоцитов. Признаки денервации на ЭМГ отсутствуют

6
Биохимически в сыворотке крови увеличено содержание саркоплазматических ферментов – креатинфосфокиназы (КФК), альдолазы, лактатдегидрогеназы и др. Эти нарушения свидетельствуют о гибели миоцитов и на 1-1,5 года опережают появление клинических симптомов
, альдолазы, лактатдегидрогеназы и др. Эти нарушения свидетельствуют о гибели миоцитов и на 1-1,5 года опережают появление клинических симптом")
7
Наиболее распространенной и злокачественной формой нервно-мышечной патологии детского возраста является прогрессирующая Х-сцепленная псевдогипертрофическая миодистрофия Дюшенна/Беккера

8
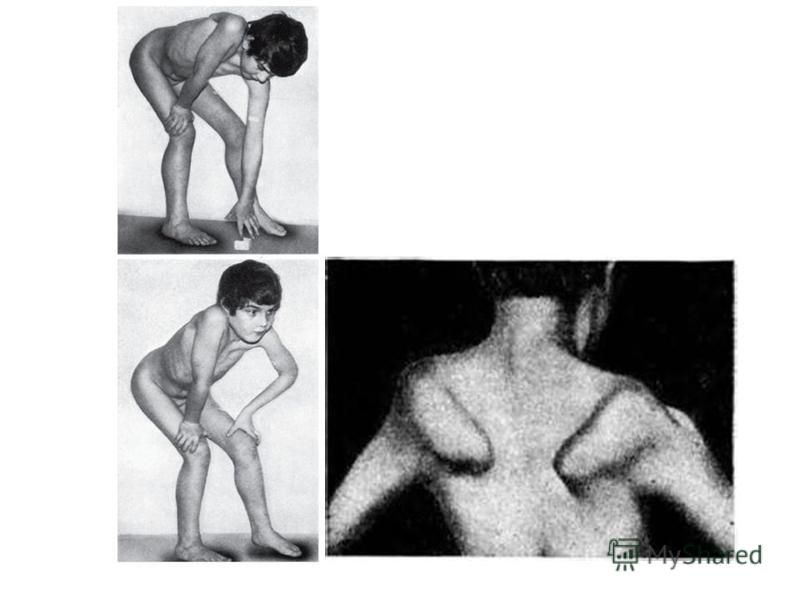
Первые признаки миодистрофии Дюшенна появляются в возрасте 2-7 лет. При начале ходьбы отмечаются неловкость в движениях, быстрая утомляемость. Постепенно появляются затруднения при подъёме по лестнице, вставании из положения на корточках, беге, ходьбе

9
Патологический процесс носит восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса и проксимальных отделов верхних конечностей. В процесс вовлекаются длинные мышцы спины, формируются поясничный гиперлордоз, «осиная талия», крыловидные лопатки, симптом свободных надплечий, «утиная походка»

11
Характерно лицо «сфинкса», «миопатия» - гипертелоризм, недостаточность мимической мускулатуры. Возникают вторичные деформации позвоночника, грудной клетки, ретракции сухожилий, контрактуры суставов. Примерно у четверти больных диагностируется олигофрения в степени дебильности

12
Больные миодистрофией Дюшенна сохраняют способность к ходьбе до летнего возраста, после чего передвигаются только с помощью инвалидной коляски. Погибают в возрасте 18 – 25 лет от интеркуррентных заболеваний, не оставляя потомства.

13
Миодистрофия Беккера – это более мягкий вариант Х- сцепленной прогрессирующей мышечной дистрофии. В настоящее время убедительно доказано, что обе формы миодистрофии обусловлены разными мутациями в одном и том же гене – DMD (Xp21.2)
")
14
В некоторых случаях дюшенна- подобные миодистрофии наследуются по аутосомно- рецессивному типу.

15
Конечностно-поясные миодистрофии – это гетерогенная группа заболеваний с преимущественной локализацией дистрофического процесса в мышцах плечевого и тазового пояса

16
При некоторых формах конечностно-поясная миодистрофия сочетается с выраженной патологией других систем, например с буллезным эпидермолизом или контрактурами суставов и кардиомиопатией - миодистрофия Эмери-Дрейфуса

17
В самостоятельную клиническую группу традиционно выделяют врожденные непрогрессирующие миопатии. Наиболее частой из них является мирозин-дефицитная миопатия. Характерной чертой врожденных миопатий Бетлема и Ульриха является их сочетание с контрактурами суставов

18
Патологические процессы при некоторых врожденных непрогрессирующих миопатиях обусловлены отложением в миофибриллах гистологически идентифицируемых аномальных образований

19
Лице-лопаточно-плечевая мышечная дистрофия Ландузи- Дежерина – третье по частоте аутосомно-доминантное заболевание мышц. Первые признаки заболевания обычно появляются во второй декаде жизни

20
Преимущественно поражается мускулатура лица, плечевого пояса и проксимальных отделов верхних конечностей. Слабость мускулатуры лица проявляется неполным смыканием век, бедностью мимики, трудностями при употреблении соломинки, сосании

21
Предполагается, что мутации, вызывающие лице-лопаточно- плечевую миодистрофию, нарушают не структуру или функцию какого-то специфического гена, но транскрипционный контроль одного или нескольких генов

22
Спинальные амиотрофии – это гетерогенная группа наследственных заболеваний, обусловленных прогрессирующим разрушением мотонейронов передних рогов спинного мозга и в некоторых случаях ствола мозга, приводящим к развитию денервационных атрофий и парезов соответствующих мышечных групп

23
Наиболее распространенной аутосомно-рецессивной формой поражения периферического двигательного неврона является проксимальная спинальная мышечная атрофия (СМА). Частота заболевания – 1 на 6-10 тысяч новорожденных
. Частота заболевания – 1 на 6-10 тысяч новорожденных")
24
Основными клиническими проявлениями СМА являются слабость и гипотония мышц, вялые симметричные парезы всей поперечно-полосатой мускулатуры, угнетение рефлексов

25
СМА делят на 3 формы: болезнь Верднига-Гоффмана, острая детская СМА I, с дебютом до 6 месяцев, хроническая, или СМА II с началом заболевания во втором полугодии жизни и СМА III, известная как болезнь Кугельберга-Веландер, дебютирует после года или позднее

26
Болезнь Верднига-Гоффмана может проявляться уже во внутриутробном периоде недостаточно активным шевелением плода, при рождении – генерализованной мышечной гипотонией («синдром шарфа», «вялый ребёнок», «поза лягушки»), в дальнейшем отставанием в моторном развитии
, в дальнейшем отставанием в мотор")
27
Развиваются вялые, преимущественно проксимальные парезы конечностей, глубокие рефлексы отсутствуют. Обычно больные погибают в течение первого года жизни от двигательной недостаточности на фоне рецидивирующих пневмоний

28
При СМА II ребёнок может научиться сидеть и даже ходить с поддержкой. При этом походка раскачивающаяся, с опорой на внутренние поверхности стоп. Затем развиваются Х-образные искривления нижних конечностей, деформации грудной клетки и позвоночника по типу кифосколиоза, появляются контрактуры в конечностях, и самостоятельное передвижение становится невозможным

29
Болезнь Кугельберга-Веландер характеризуется более медленным течением сходных патологических процессов на протяжении десятилетий. Продолжительность жизни при СМА II и СМА III может достигать 20 и 30 лет соответственно

30
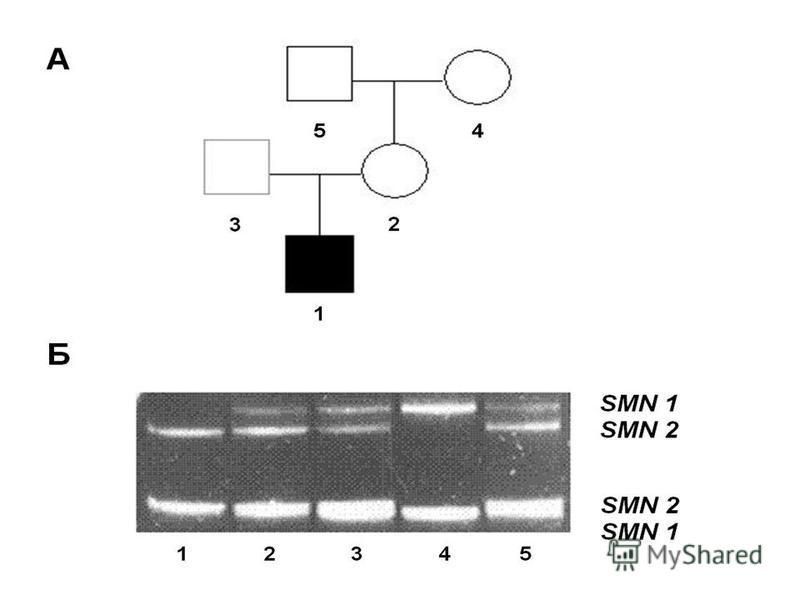
Все клинические типы СМА обусловлены мутациями в гене SMN1 (5q13.2), кодирующем белок выживания двигательных нейронов – Smn-белок. От 95% до 98% больных СМА имеют гомозиготные делеции различной протяженности, затрагивающие экзоны 7 и 8 гена SMN1
, кодирующем белок выживания двигательных нейронов – Smn-белок. От 95% до 98% больных СМА имеют гомозиготные делеции различной протяженности, затрагивающие экзоны 7 и 8 гена SMN1")
32
Наследственные полиневропатии

33
Наследственные полиневропатии составляют до % всех хронических полиневропатий и представляют собой весьма гетерогенную группу. Наиболее распространёнными являются наследственные моторно- сенсорные невропатии, или болезнь Шарко-Мари-Тута

34
Клинически характеризуются прогрессирующей слабостью и атрофией дистальной (преимущественно перонеальной) мускулатуры; расстройствами чувствительности по полиневритическому типу; деформацией стоп типа Фридрейха; расстройством походки типа «степпаж» (перонеальная, петушиная походка, обусловленная отвислой стопой)
 мускулатуры; расстройствами чувствительности по полиневритическому типу; деформацией стоп типа Фридрейха; расстройством походки типа «степпаж» (")
35
Патогенетически моторно-сенсорная невропатия делится на 2 основных типа: демиелинизирующая – миелопатия и аксональная– аксонопатия. Для первых характерно снижение скорости проведения импульса по нерву, морфологически сегментарная гипертрофическая демиелинизация с образованием «луковичных» головок

36
Для второго типа характерно первичное поражение аксонов, нормальная скорость проведения импульса, морфологически сохранность структуры миелина

37
Из сравнительно редких синдромов, отличающихся от классического фенотипа Шарко-Мари-Тута следует отметить синдром Дежерина-Сотта, основными клиническими проявлениями которого являются выраженные гипертрофии периферических нервов («гипертрофический неврит»), снижение скорости проведения импульса по двигательным волокнам, ранняя инвалидизация

38
Синдром Дежерина-Сотта также генетически гетерогенен, но не является самостоятельной формой, а представляет собой аллельные варианты различных генетических типов болезни Шарко-Мари-Тута

39
Наследственные миотонии и миоплегии (нервно-мышечные каналопатии)
")
40
Феномен миотонии заключается во внезапном тоническом спазме мышцы, возникающем вслед за произвольным её сокращением. Он может застать больного в любое время – при выполнении рабочих операций, еде, переходе улицы и т.п.

41
Известны две клинические формы: аутосомно-доминантная врождённая миотония Томсона и аутосомно- рецессивная генерализованная миотония Беккера. Характерной особенностью миотонии Томсона является атлетическое телосложение. Форма Беккера более тяжёлая и обычно сопровождается постоянной мышечной слабостью

42
Обе формы обусловлены мутациями в гене хлорного канала скелетных мышц – CLC1 (7q34). Известно, что ионы хлора обеспечивают стабилизацию мембранного покоя после акта сокращения
. Известно, что ионы хлора обеспечивают стабилизацию мембранного покоя после акта сокращения")
43
Наследственная пароксизмальная миоплегия характеризуется приступами резкой слабости, вплоть до полного паралича рук и ног. Выделяют 3 формы заболевания: гипо-, гипер- и нормокальциемический периодический паралич

44
Аутосомно-доминантная гипокалиемическая форма, или болезнь Шахновича-Вестфаля встречается наиболее часто. Приступы появляются с детства, от единичных до каждодневных, чаще утром, при этом больные просыпаются обездвиженными

45
Краниальная мускулатура, как правило, не страдает, сознание сохранено. Длительность приступа от 30 минут до 72 часов, он может провоцироваться обильным приёмом пищи, физической нагрузкой. В момент приступа уровень сывороточного калия снижается до 2 ммоль/л и ниже

46
Гиперкалиемическая форма пароксизмальной миоплегии, или болезнь Гармстропа отличается слабостью мимической и артикуляционной мускулатуры, приступ может провоцироваться отдыхом, голоданием. Третья, очень редкая нормокальциемическая миоплегия по клиническим проявлениям не отличается от предыдущих форм

47
Наследственные миастении

48
В разделе наследственных нейромышечных заболеваний традиционно рассматривают миастению, или болезнь Эрба. Это многофакторное аутоиммунное заболевание, относящееся к болезням нейромышечного синапса

49
Миастения клинически характеризуется нарастающей при выполнении движений мышечной слабостью, которая может достигать степени паралича. После отдыха объём движений увеличивается и даже достигает нормы. Как правило, атрофии и расстройства чувствительности отсутствуют

50
Различают миастению генерализованную и локальную с поражением глазодвигательных нервов (глазная форма) и мышц гортани, глотки, языка (бульбарная форма). В зависимости от формы ведущими клиническими проявлениями являются двоение в глазах, птоз, дизартрия, дисфагия, общая слабость. Эти симптомы меняются в течение суток и зависят от физической нагрузки
 и мышц гортани, глотки, языка (бульбарная форма). В зависимости от формы ведущими клиническими проявлениями являются двоение в глазах, птоз, дизарт")
51
Опасны миастенические кризы, когда внезапно развиваются генерализованная общая слабость и выраженные бульбарные расстройства, нарушения дыхания, требующие применения аппаратных методов

52
В патогенезе аутоиммунной реакции основную роль играет тимус. При патологии тимоцитов (тимома, воспаление и др.) их антигенный состав становится сходным с антигенами ацетилхолиновых рецепторов, рецепторами Са-каналов. В результате начинают продуцироваться аутоантитела, приводящие к гибели ацетилхолиновых рецепторов
 их антигенный состав становится сходным с антигенами ацетилхолиновых рецепторов, рецепторами Са-каналов. В результате начинают продуциров")
53
Пресинаптический миастенически синдром, сопровождающийся эпизодической атаксией, обусловлен мутациями в гене CHAT (10q11.23), кодирующем холинацетилтрансферазу – биосинтетический фермент для нейротрансмиттера ацетилхолина
, кодирующем холинацетилтрансферазу – биосинтетический фермент для нейротрансмиттера ацетилхолина")
Еще похожие презентации в нашем архиве:

 Работа студента 309 л группы Комарова К. В. МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ ФГАОУ.")




 описан Фукухама в 1980 г.")
. Мукополисахаридоз II типа (МПС II, OMIM - 309900), или синдром Хантера - это лизосомная болезнь накопления.")

 – 14. Преподователь: Козуев Кадыр.")




 группа генетических нарушений. Одно из заболеваний ломкости костей. Люди с НО либо имеют недостаточное.")





