Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

2
группа наследственных тубулопатий, клиническая картина которых в ранние сроки заболевания имитирует рахит, но не связана с дефицитом поступающего в организм витамина D; их ведущим синдромом являются аномалии скелета (почечные остеопатии).
.")
3
Патогенетические механизмы формирования первичных (наследственных) тубулопатий: Генетически детерминированные нарушения структуры мембранных белков-носителей; Энзимопатии (наследственно обусловленная недостаточность ферментов, обеспечивающих активный мембранный транспорт); Изменения чувствительности рецепторов клеток канальцевого эпителия к действию гормонов; Изменения общей структуры цитомембран клеток при дисплазиях, в происхождении которых определенная роль принадлежит наследственным факторам. Вторичные тубулопатий возникают: в результате повреждения транспортных систем почечных канальцев как при наследственных, так и при приобретенных болезнях обмена в связи с нарушениями метаболизма за пределами нефрона.
 тубулопатий: Генетически детерминированные нарушения структуры мембранных белков-носителей; Энзимопатии (наследственно обусловленная недостаточность ферментов, обеспечивающих активный")
4
витамин-D- резистентный рахит витамин-D-зависимый рахит болезнь де Тони Дебре Фанкони почечный тубулярный ацидоз.

5
с первичным нарушением процессов всасывания кальция и фосфора в кишечнике; с первичным дефектом транспорта неорганических фосфатов в почках и повышением чувствительности эпителия канальцев почек к действию паратгормона; с генетически детерминированным сочетанием этих нарушений; с синтезом в организме фосфатурических метаболитов витамина D и недостаточным образованием 25-оксихолекальциферола в печени.

6
задержка роста выраженные прогрессирующие деформации скелета, особенно нижних конечностей (по варусному типу, что сопровождается нарушением походки ребенка («утиная походка»); значительная болезненность костей и мышц, нередко мышечная гипотония;
; значительная болезненность костей и мышц, нередко мышечная гипотония;")
7
Б/х крови гипофосфатемия, гиперфосфатурия, нормальные показатели общего кальция в крови (2,32,75 ммоль/л), повышение активности щелочной фосфатазы крови, повышение уровня паратгормона в крови, снижение абсорбции кальция и фосфора в кишечнике; Рентгенологическое исследование: выявляют два типа костных изменений: 1)изменения эпифизов костей с расширением зон пролиферации хряща, грубоволокнистую структуру кости с признаками остеоидной гиперплазии (полная аналогия с рахитом), 2)поражение метафизов уже сформировавшейся кости и проявления остеомаляции. Гистологическое исследование костной ткани - нарушение структуры костных каналов и трабекул, пролиферацию хряща, чередование усиленного образования остеоидной ткани с участками остеопороза.
, повышение активности щелочной фосфатазы крови, повышение уровня паратгормона в крови, снижение абсорбции кальция и фосфора в кишечнике; Рентген")
8
В активной фазе болезни при наличии болей в костях и суставах показан постельный режим продолжительностью до 2 нед. Основными препаратами в терапии являются витамин D и его метаболиты.

9
Начальные дозы витамина D составляют ЕД в сутки. Их увеличивают под контролем показателей кальция и фосфора в сыворотке крови и моче, активности щелочной фосфатазы крови, исследования уровня которых должно проводиться каждые 1014 дней. Максимальные суточные дозы витамина D в зависимости от клинико-биохимических вариантов болезни составляют: при первом варианте ЕД, при втором ЕД, при третьем ЕД. При четвертом варианте болезни назначение витамина D противопоказано.

10
Препараты кальция: глюконат кальция или хлорид кальция по 1,52 г в сутки) и фосфора (фитин 11,5 г в сутки, глицерофосфат кальция 0,51 г в сутки). Для улучшения процессов всасывания кальция и фосфора в кишечнике рекомендуют длительное (56 мес.) применение концентрированных цитратных смесей (например, лимонная кислота 24 г, цитрат натрия 48 г и дистиллированная вода 500 мл) по 2050 мл в сутки.
 и фосфора (фитин 11,5 г в сутки, глицерофосфат кальция 0,51 г в сутки). Для улучшения процессов всасывания кальция и фосфора в кишечнике рекомендуют длительное (56 мес.) примен")
11
улучшение общего состояния больных, увеличение темпов роста детей, нормализация или значительное улучшение показателей фосфорно-кальциевого обмена, снижение активности щелочной фосфатазы крови положительная динамика структурных изменений костной ткани (по данным рентгенологического исследования).

12
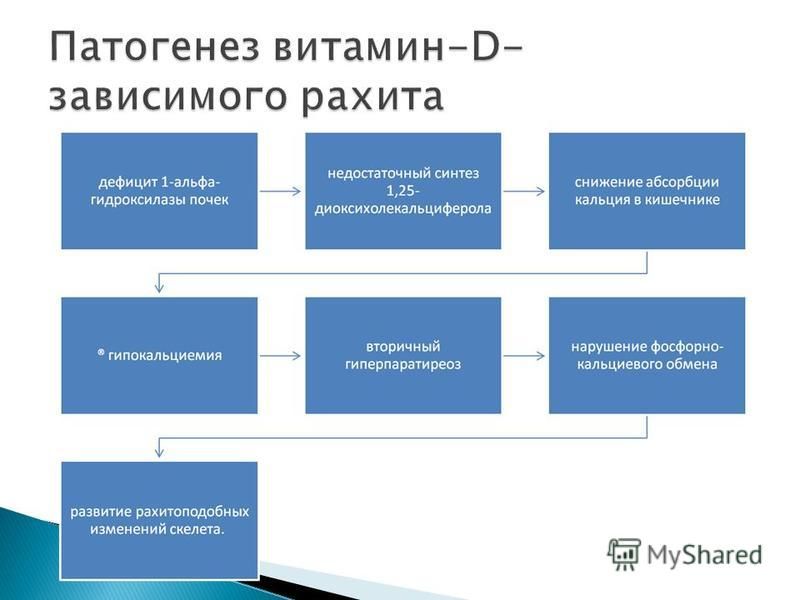
энзимопатия, связанная с дефектом фермента 1-альфа-гидроксилазы в почках, осуществляющего превращение 25- оксихолекальциферола в 1,25- диоксихолекальциферол. Имеет аутосомно-рецессивный тип наследования, однако встречаются и спорадические случаи заболевания

14
Наследуется по аутосомно- рецессивному типу, однако экспрессивность мутантного гена в гомозиготном состоянии значительно варьирует; встречаются спорадические случаи, обусловленные свежей мутацией. Первые признаки заболевания появляются во второй половине первого года жизни, развернутый симптомокомплекс формируется ко второму году жизни; реже наблюдается поздняя манифестация болезни в 67-летнем возрасте.

16
Начальные клинические проявления: повышенная жажда, полиурия, иногда длительный субфебрилитет, рвота. На втором году жизни выявляют отставание физического развития и костные деформации нижних конечностей (вальгусные или варусные), грудной клетки, предплечий и плечевых костей.
, грудной клетки, пре")
17
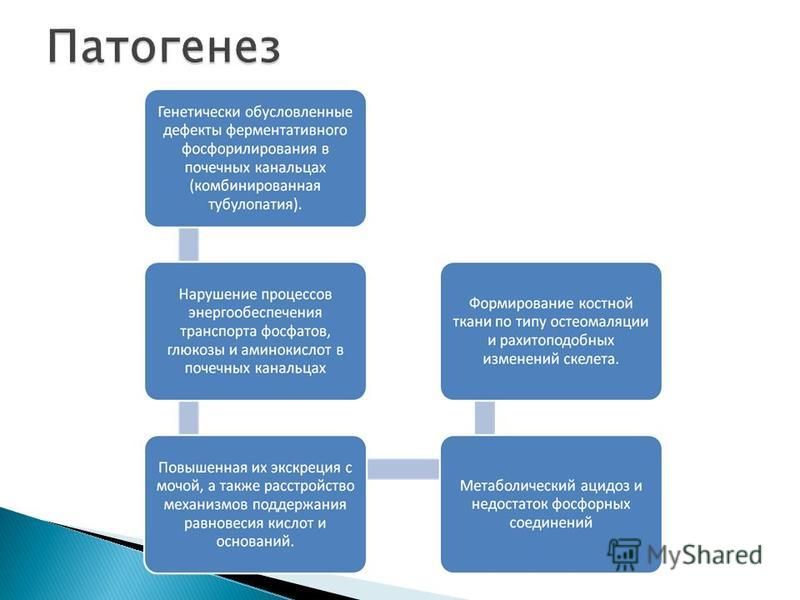
Б/х крови снижение уровня кальция и фосфора, повышение активности щелочной фосфатазы, метаболический ацидоз (рН 7,357,25; ВЕ = 1012 ммоль/л). Б/х мочи Экскреция кальция с мочой обычно остается нормальной при повышенном клиренсе фосфатов мочи. глюкозурия (2030 г/л и выше), генерализованная гипераминоацидурия нарушение функций аммониоацидогенеза снижение титрационной кислотности, повышение рН мочи > 6,0 При полиурии до 2 л и более в сутки удельная плотность мочи, как правило, высокая ( ), что обычно связано с глюкозурией. Рентгенологически при этом определяют остеопороз системный различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от биологического возраста ребенка.
. Б/х мочи Экскреция кальция с мочой обычно остается нормальной при повышенном клиренсе фосфатов мочи. глюкозу")
18
I вариант характеризуется значительной задержкой физического развития, тяжелым течением заболевания с выраженными костными деформациями и нередко переломами костей, резкой гипокальциемией (1,61,8 ммоль/л), снижением абсорбции кальция в кишечнике. II вариант отмечают умеренную задержку физического развития, легкое течение с незначительными костными деформациями, нормокальциемию и нормальное усвоение кальция в кишечнике.
, снижением абсорбции кальция в кишечнике. II")
Еще похожие презентации в нашем архиве:




 – ферменты, гидролизирующие эфиры фосфорной кислоты. В зависимости от значения рН, при котором действует фермент, различают:")
 – это синдром необратимого нарушения функции почек, которое наблюдается.")

. Мукополисахаридоз II типа (МПС II, OMIM - 309900), или синдром Хантера - это лизосомная болезнь накопления.")

 из них : 99% - костной и зубной тканях, в форме гидроксиапатита 3 Ca 3 (PO 4 ) 2 * Ca(OH) 2 фторапатита.")


 описан Фукухама в 1980 г.")



. В основе.")



