Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
Мышечная дистрофия Дюшенна-Беккера Подготовил студент Группы ВИ-512 Рябущенко Дмитрий

2
Код по OMIM Код по МКБ-10 - G71.0

3
Наследственное заболевание с Х-сце- пленным рецессивным типом наследования, обусловленное мутацией гена, кодирующего белок миоцитов-дистрофия.

4
Наследуется по рецессивному типу, сцепленному с X-хромосомой Частота встречаемости - составляет около 30 на новорожденных мальчиков Частота встречаемости - у 1 человека на 4000 новорожденных мужского пола. Мутации в гене дистрофия могут быть унаследованы или возникают спонтанно во время зародышевой линии передачи.

5
Каждый сын, рожденный женщиной с мутацией в гене дистрофияа в одной из двух ее Х- хромосом, имеет 50% вероятность унаследовать поврежденный ген и иметь МДД. Каждая дочь такой женщины имеет 50% вероятность унаследовать мутацию и стать носительницей. У носительниц обычно нет симптомов заболевания, но у них могут быть дети с мутацией или заболеванием.

6
Мышечная дистрофия Дюшенна обусловлена мутацией в гене дистрофия, локус которого Xp21. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофияа приводит к проникновению избыточного кальция в сарколемму (клеточную мембрану). Как следствие изменения этих сигнальных путей, вода наполняет митохондрии, которые после этого разрываются. При дистрофии скелетных мышц, митохондриальная дисфункция приводит к усилению стресса вызванного цитозольным-кальциевым сигналом и усилению производства стресс-индуцированных активных форм кислорода (АФК). В этом сложном каскадном комплексе, который включает в себя несколько реакций еще до сих пор не понятно до конца, почему из-за повреждения сарколеммы увеличиваются проявления окислительного стресса, который в итоге приводит к смерти клетки. Мышечные волокна подвергаются некрозу и, наконец, происходит замена мышечной ткани жировой, а также соединительной.
 через белковый комплекс, который со")
9
Первые симптомы появляются в 3 – 5 – 6 лет

10
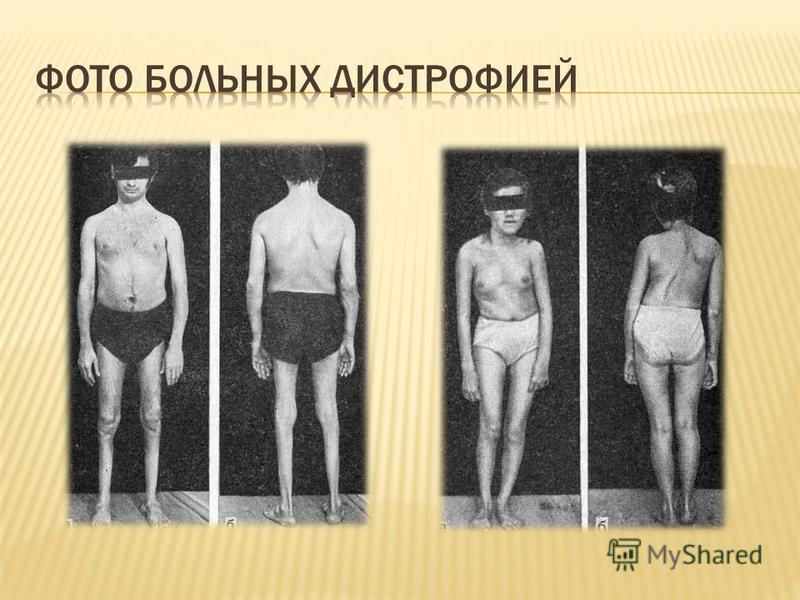
Мышечная слабость, которая в первую очередь связана с атрофией мышц, а именно скелетной мышечной ткани. В первую очередь атрофируются мышцы бедер, таза, плеч и икроножные мышцы. Мышечная слабость возникает также в руках, шее и других частях тела, но обычно не так рано, как в нижней части тела. Псевдогипертрофия Икроножных мышц

11
Мальчики часто падают,отстают в играх от сверстников, с трудом бегают и прыгают Симптом лестницы - к 5 годам мышечная слабость выявляется при осмотре. Из положения сидя на полу больной встает, опираясь сначала на собственные колени, затем на бедра Как правило, утолщены голени, причем истинная гипертрофия икроножных мышц в начале болезни со временем сменяется псевдо гипертрофией - мышца замещается жировой и соединительной тканью К 6 годам формируются контрактуры ахилловых сухожилий и подвздошно- большеберцовых трактов Изменена походка - на цыпочках, с переразгибанием в поясничном отделе позвоночника. Мышечная слабость нарастает, преимущественно страдают проксимальные мышцы ног (особенно) и рук, сгибатели шеи С 8-10 лет больным требуются костыли, из-за преимущественно сидячего положения усиливаются контрактуры и ограничиваются движения в тазобедренных, коленных, локтевых, лучезапястных суставах К 12 годам больные прикованы к коляске Контрактуры становятся необратимыми, часто возникает и прогрессирует сколиоз, вызывающий боли От этого деформируется грудная клетка и ухудшается функция легких, которая и без того страдает из-за мышечной слабости В лет часто развиваются тяжелые пневмонии, нередко с летальным исходом

13
У всех больных активность КФК в сыворотке повышена в раз. Она высока уже с рождения, а при далеко зашедшей болезни снижается из-за обездвиженности и уменьшения мышечной массы. При ЭМГ выявляется миопатическая триада - снижение амплитуды и длительности и полифазные (более чем из четырех фаз) потенциалы действия двигательных единиц; количество двигательных единиц, вовлекающихся при произвольном сокращении, возрастает.

14
ДНК-тест - причиной миопатии Дюшенна является дефект дистрофияа - белка с молекулярной массой , находящегося на внутренней поверхности сарколеммы Ген дистрофияа - один из самых крупных идентифицированных генов человека, его длина - 2 млн нуклеотидов. Он находится в сегменте Хр 21 В настоящее время известные мутации удается выявить примерно у двух третей больных Делеции располагаются по длине гена неравномерно, чаще в его начале (5'-концевая область) и в середине Течение болезни не зависит от размера делеции. Реже встречаются дупликации гена и точечные мутации Наличие специфичной мутации однозначно подтверждает диагноз и позволяет надежно выявлять носительниц мутации.

15
При биопсии видны разнокалиберные мышечные волокна, а также небольшие группы некротизированных и регенерирующих волокон; большинство мышечных волокон заменены соединительной и жировой тканью. Несомненным подтверждением диагноза служит выявление в биоптате недостаточности дистрофияа либо обнаружение мутации молекулярно- генетическими методами.

16
- употребление таких кортикостероидов как преднизолон и дефлазакорт - рандомизированные контролируемые исследования показывают, что использование бета 2-агонистов увеличивает мышечную силу, но не замедляет процесс прогрессирования заболевания (время контроля 12 месяцев) - рекомендуется умеренная физическая активность, разрешается заниматься плаванием - для поддержания мышечной силы, гибкости и функциональности суставов важна физиотерапия; - использование ортопедических приспособлений - по мере прогрессирования заболевания необходимым становится использование специальных респираторных механизмов, позволяющих обеспечить нормальный процесс дыхания

17
Больные МДД, как правило, живут только к подростковому возрасту или умирают в возрасте лет. Последние достижения в области медицины, позволяют надеяться на увеличение продолжительности жизни больных этим расстройством. Иногда (но очень редко) особи с МДД доживали до лет
 о")
Еще похожие презентации в нашем архиве:






. Мукополисахаридоз II типа (МПС II, OMIM - 309900), или синдром Хантера - это лизосомная болезнь накопления.")

 описан Фукухама в 1980 г.")











 служит опорой телу и его органам. Состоит из костей черепа,")

