Скачать презентацию
2
boli determinate de lipsa uneia, sau a mai multor enzime lizozomale - reprezintă enzimopatii ereditare - boli determinate de lipsa uneia, sau a mai multor enzime lizozomale; se manifesta prin acumulare treptata in lizozomi a substratului enzimei lipsa; - se manifesta prin acumulare treptata in lizozomi a substratului enzimei lipsa; -se caracterizează cu debut la nou- născut şi majoritatea se manifestă printr-o patologie neurologică şi un handicap sever; -se transmit autozomal – recesiv; - se întâlnesc cu o frecvenţă de 1:2500 nou-născuţi. Ce este o boala lizozomala???

3
Biogeneza lizozomilor

4
Ac umularea substratului enzimei lipsa

5
ETIO-PATOGENIA BOLILOR LIZOSOMALE MUTAŢIE în gena EL(x) DEFICITUL UNEI ENZIME DIN LIZOSOM ACUMULAREA SUBSTRATULUI şi încărcarea celulei cu substrat toxic Semne şi simptome specifice ale bolii dependente de tipul enzimei, substratului şi celulei afectate
 DEFICITUL UNEI ENZIME DIN LIZOSOM ACUMULAREA SUBSTRATULUI şi încărcarea celulei cu substrat toxic Semne şi simptome specifice ale bolii dependente de tipul enzimei, substratului şi celulei afect")
6
Maladiile lizozomale (boli de stocaj) se clasifica in: Sfingolipidoze(SF) Sfingolipidoze(SF) Boala Gaucher Boala Gaucher Boala Wolman Mucolipidoze (ML) Mucolipidoze (ML) Mucolipidoza de tip II, Mucolipidoza de tip II, Boala Pompe Mucopolizaharidoze (MPZ) Mucopolizaharidoze (MPZ) Tip I Sindromul Hunter Tip I Sindromul Hunter Tip II Tip II Sindromul Hurler
 se clasifica in: Sfingolipidoze(SF) Sfingolipidoze(SF) Boala Gaucher Boala Gaucher Boala Wolman Mucolipidoze (ML) Mucolipidoze (ML) Mucolipidoza de tip II, Mucolipidoza de tip II, Boala Pompe Mucopolizaharidoze (")
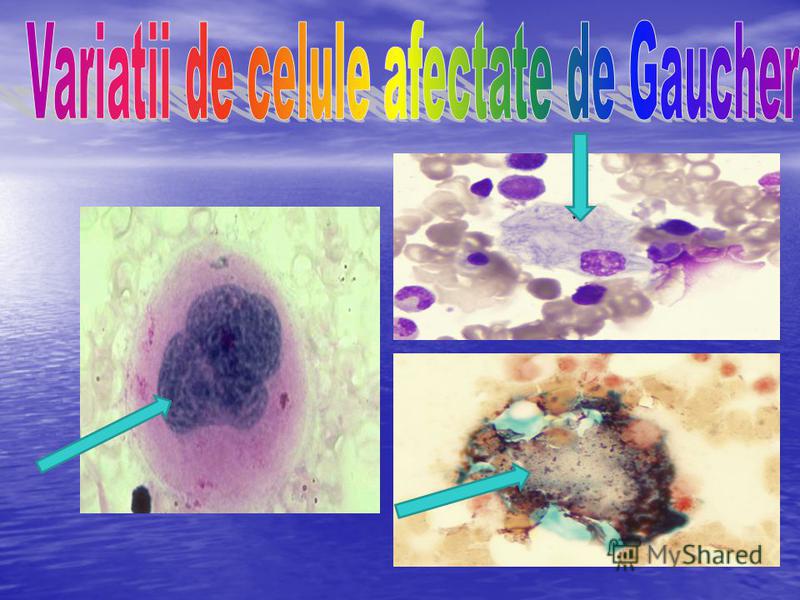
7
Sfingolipidozele - se datoresc degradării incomplete a gangliozidelor, sfingomielinei, sau a glicosfingolipidelor, la nivel lizozomal. *fregventa 1: nou – născuţi *CAUZA- o serie de mutaţii la nivelul genei GBA, localizată 1q21 *Boala se manifestă prin: -splenomegalie-hepatomegalie-osteopatia

8
Deficit enzimatic: deficitul de b-glucozidază Frecventa 1: Transmitere - autosomal recesiv crs. 1q21 Date clinice si de laborator: Forma acuta neuropatica - retard mental - hepatosplenomegalie acumulare de glucocerebrosid in lizozomii sistemului RE si SNC - moartea se produce in 1-2 ani Forma cronica -mai frecventa la evreii est-europeni -Hepatosplenomegalie - leziuni osteolitice

10
Gaucher de tip 1 Caracteristica: Caracteristica: Este mai frecventă la persoanele de origine evreiască şi nu afectează sistemul nervos central. Este mai frecventă la persoanele de origine evreiască şi nu afectează sistemul nervos central. Hepatosplenomegalie. Hepatosplenomegalie. Dureri osoase acute, insotite de fracturi fregvente. Dureri osoase acute, insotite de fracturi fregvente. Anemie cauzată de un nivel scăzut de fier in eritrocite. Anemie cauzată de un nivel scăzut de fier in eritrocite.

11
GAUCHER DE TIP 2 Caracteristica: Caracteristica: Incidenţa este foarte scăzuta. Incidenţa este foarte scăzuta. Cea mai severă formă de prezentare fenotipica. Cea mai severă formă de prezentare fenotipica. Anomalii neurologice congenitale. Anomalii neurologice congenitale. Deteriorarea progresivă a creierului. Deteriorarea progresivă a creierului. Este de obicei, cauzele de deces în primii doi ani de viaţă. Este de obicei, cauzele de deces în primii doi ani de viaţă.

12
Boala Wolman Boala Wolman - se datoreşte insuficienţei de lipază acidă Hepatocite şi histiocite de diferite mărimi cu vacuole

13
Deficit enzimatic: lipază acidă Transmitere - autosomal recesiv Date clinice si de laborator: -debut in copilarie -simptome gastro-intestinale -hepatosplenomegalie -acumularea de esteri ai colesterolului si trigliceride in lizozomii sistemului RE -moarte in 3-6 luni –

14
Mucolipidozele (ML) Mucolipidozele (ML) - sunt un grup de boli, în care, la nivel lizozomal, se depun concomitent, MPZ şi lipide.
 Mucolipidozele (ML) - sunt un grup de boli, în care, la nivel lizozomal, se depun concomitent, MPZ şi lipide.")
15
Mucolipidoza de tip II La nivelul celulelor hepatice se acumulează acid sialic, precum şi lipide neutre. Există şi o uşoară vacuolare a hepatocitelor.

16
Deficit enzimatic: N-acetil-glucosamin-fosfotransferaza Transmitere - autosomal recesiv Date clinice si de laborator: -r-retard mental -s-statura foarte mica - modificari scheletale -t-trasaturi grosiere faciale

17
Boala Pompe Glicogenoza tip II (Pompe) Boala PompeBoala Pompe – determinată de insuficienţei de maltază acidă. Toate celulele ficatului participă la stocare, luând un aspect vacuolizat.
 Boala PompeBoala Pompe – determinată de insuficienţei de maltază acidă. Toate celulele ficatului participă la stocare, luând un aspect vacuolizat.")
18
A fost descrisa pentru prima data de J.C. Pompe in anul 1932 la un copil de 7 ani ca o hipertrofie cardiaca acuta. Frecvenţa medie este de 1/ de locuitori. Deficit de alfa-1-glucozidaza Gena α-glucozidazei (localizată pe crs. 17 q25.2-q formata din 20 exoni) Se transmite autozomal recesiv

19
Forme clinice Forma clasică (generalizată, infantilă) Debut precoce la varsta de sugar, caracterizat prin: cardiomegalie hepatomegalie hipotonie insuficienţă cardio-respiratorie acuta. Debut dupa virsta de sugar: Afectarea musculaturii scheletice. Insuficienţă respiratorie. Invaliditate - prin afectarea muscularei proximale.
 Debut precoce la varsta de sugar, caracterizat prin: cardiomegalie hepatomegalie hipotonie insuficienţă cardio-respiratorie acuta. Debut dupa virsta de sugar: Afectarea musculaturii scheletice. In")
20
Fenotipul bolii Pompe Acumularea de glicogen este masivă la nivelul cordului, musculaturii scheletice şi în ficat în forma generalizată şi este mai redusă şi limitată deobicei la musculatura scheletică în forma cu debut tardiv.

21
Mucopolizaharidozele (MPZ) - boli datorate insuficienţei unor enzime ce participă la catabolismul glicozaminoglicanilor. Mucopolizaharidozele (MPZ) - boli datorate insuficienţei unor enzime ce participă la catabolismul glicozaminoglicanilor. Mucopolizaharidoze (MPZ) Tip I Mucopolizaharidoze (MPZ) Tip I Tip II Tip II
 - boli datorate insuficienţei unor enzime ce participă la catabolismul glicozaminoglicanilor. Mucopolizaharidozele (MPZ) - boli datorate insuficienţei unor enzime ce participă la catabolismul glicozaminoglicanilor. Mucopoli")
22
S. Hunter S. Hunter Deficit de ioduronidaza 2 sulfataza Fregventa 1: de nou-nascuti Degradare mentala Malformatii congenitale multiple

23
Deficit enzimatic: ioduronidaza 2 sulfataza Transmitere - X -linkat recesiv Date clinice si de laborator: -- hepatoslenomegalie -retard mintal variabil -opacitaţi corneene -Cifoza

25
Sindromul Hurler -insuficienţa de alfa-L-iduronidază Hepatocite tumefiate şi vacuolate Sindromul Hurler -insuficienţa de alfa-L-iduronidază Hepatocite tumefiate şi vacuolate

26
Deficit enzimatic de a-iduronidaza FREGVENTA: 1: DE NOU-NASCUTI -Boala autosomal recesiva. Date clinice si de laborator: -Retard mental. -Hepatoslenomegalie -Insuficienta cardiaca grava

27
Variatii de celule afectate de Hurler

28
Gena responsabila Gena responsabila GENA IUAD GENA IUAD –Locus: Cromozomul 4p16.3 4p16.3 Cauza bolii Hurler – Cauza bolii Hurler – insertia sau deletia nucleotidelor la nivelul genei IUAD. insertia sau deletia nucleotidelor la nivelul genei IUAD.

29
Functia Genei IUAD Functia Genei IUAD La nivel molecular – sinteza alpha-L-iduronidazei ; sinteza alpha-L-iduronidazei ; La nivel celular – Asigura posibilitatea functionarii normale a lizozomilor. La nivel celular – Asigura posibilitatea functionarii normale a lizozomilor. La nivel organismic- La nivel organismic- Metabolizmul normal al mucopolizaharidozelor. Metabolizmul normal al mucopolizaharidozelor.

30
Evolutia bolii Hurler Evolutia bolii Hurler In primul an de viata: In primul an de viata: opacitatea corneei opacitatea corneei In al doilea an de viata: In al doilea an de viata: Probleme cardiace; Probleme cardiace; Hipertensiune; Hipertensiune; Infectii otorinolaringolice si pulmonare ; Infectii otorinolaringolice si pulmonare ; Tulburari de somn(insomnie); Tulburari de somn(insomnie); Represii psiho-motorice; Represii psiho-motorice;

31
BOALA SE PREZINTA PRIN DOUA FORME : BOALA SE PREZINTA PRIN DOUA FORME : FORMA ACUTA FORMA ACUTA FORMA ATENUATA FORMA ATENUATA

32
Simptome caracteristice bolii HURLER Simptome caracteristice bolii HURLER Macrocefalie Macrocefalie Fata grosiera Fata grosiera Nas aplatizat Nas aplatizat Ochi proeminenti Ochi proeminenti Limba voluminoasa: gura nu se inchidde complect. Limba voluminoasa: gura nu se inchidde complect. Abdomen proeminent. Abdomen proeminent. Prezenta numerosilor peri pe suprafata intregului corp; Prezenta numerosilor peri pe suprafata intregului corp; Diminuarea capacitatii de a face miscari; Diminuarea capacitatii de a face miscari; Deformarea de forma a miinilor. Deformarea de forma a miinilor. Malformatii ale picioarelor. Malformatii ale picioarelor. Marire in volum a ficatului si a splinei. Marire in volum a ficatului si a splinei. Tulburari de crestere: are loc retinerea in crestere si la virsta de 3 ani rareori bolnavul poate atinge 1,10m. Tulburari de crestere: are loc retinerea in crestere si la virsta de 3 ani rareori bolnavul poate atinge 1,10m.

34
Malformatii congenitale multiple

35
Concluzii Bolile lizozomale: Concluzii Bolile lizozomale: 1. Sunt cauzate de defectul unei gene ce codifica o enzima lizozomala; 1. Sunt cauzate de defectul unei gene ce codifica o enzima lizozomala; 2.Sunt boli de acumulare; 2.Sunt boli de acumulare; 3.Manifestarea clinica variată: 3.Manifestarea clinica variată: - cu evolutie progresiva; - cu evolutie progresiva; - cu afectare multisistematica: - cu afectare multisistematica: sistemul nervos sistemul nervos visceromegalie visceromegalie retard de crestere. retard de crestere.

37
Datorită h eterogenităţii alelice: Datorită h eterogenităţii alelice: mutatie la nivelul A (2134) FORMA GRAVA mutatie la nivelul A (2134) FORMA GRAVA mutatie la nivelul G (2430) FORMA MAI USOARA mutatie la nivelul G (2430) FORMA MAI USOARA Dependenta de virsta: Dependenta de virsta: odata cu inaintare in virsta pacientul achiziţionează noi simptoame odata cu inaintare in virsta pacientul achiziţionează noi simptoame odata cu inaintare in virsta boala se agraveaza odata cu inaintare in virsta boala se agraveaza ??? Interactiuni genice ??? Interactiuni genice pacienţii manifestă forme mai grave şi forme uşoare a bolii pacienţii manifestă forme mai grave şi forme uşoare a bolii ? cai alternative de metabolism a substratului dat… ? cai alternative de metabolism a substratului dat… Influenta factorilor de mediu Influenta factorilor de mediu progresarea bolii depinde de alimentatie progresarea bolii depinde de alimentatie Expresivitatea variabila a genelor patologice: progresarea bolii depinde de diferiţi factori ai mediului
 FORMA GRAVA mutatie la nivelul A (2134) FORMA GRAVA mutatie la nivelul G (2430) FORMA MAI USOARA mutatie la nivelul G (2430) FORMA MAI USOARA Dependenta")
38
Metode de diagnostic Metode de diagnostic Examinările clinice şi paraclinice: Examinările clinice şi paraclinice: examen hematologic periferic; examen hematologic periferic; teste funcţionale hepatice; teste funcţionale hepatice; radiografii osoase; radiografii osoase; ecografie abdominală; +/- TC, RMN– pentru evaluarea spleno- hepatomegaliei ecografie abdominală; +/- TC, RMN– pentru evaluarea spleno- hepatomegaliei RMN, osteodensitometrie – pentru evaluarea bolii osoase RMN, osteodensitometrie – pentru evaluarea bolii osoase

39
Metode de Tratament Tratament simptomatic Tratament simptomatic asigura tratarea simptomelor locale asigura tratarea simptomelor locale Dieta Dieta * alimentatie prin evitare de substrat * alimentatie prin evitare de substrat a enzimei lipsa a enzimei lipsa Terapie enzimatica Terapie enzimatica administrarea regulata a enzimei lipsa obtinuta pe calea ingineriei genice administrarea regulata a enzimei lipsa obtinuta pe calea ingineriei genice

40
Prevenirea – testarea prenatala 1.Diagnostic prenatal molecular 1.Diagnostic prenatal molecular studiul posibililor mutatii la nivel de genă implicată studiul posibililor mutatii la nivel de genă implicată 2.Diagnosticul prenatal poate fi stabilit şi prin dozarea β-glucozidazei acide în amniocite, obţinute prin amniocenteză în săptămâna XVI de gestaţie. 2.Diagnosticul prenatal poate fi stabilit şi prin dozarea β-glucozidazei acide în amniocite, obţinute prin amniocenteză în săptămâna XVI de gestaţie. 3. Sfatul genetic pentru calcularea riscului in cazul in care parintii sunt purtatori, sau in aceasta familie deja este un copil bolnav 3. Sfatul genetic pentru calcularea riscului in cazul in care parintii sunt purtatori, sau in aceasta familie deja este un copil bolnav

41
Terapia genica un vis sau o realitate?! Terapia genica un vis sau o realitate?! Speranţa terapeutică în perspectivă o reprezintă introducerea genei normale în organismul bolnavilor (terapia genică). Speranţa terapeutică în perspectivă o reprezintă introducerea genei normale în organismul bolnavilor (terapia genică). In prezent sunt multe incercari in acest sens ce pe viitor sa speram ca vor fi realizate !!!!!! In prezent sunt multe incercari in acest sens ce pe viitor sa speram ca vor fi realizate !!!!!!
. Speranţa terapeutică în perspectivă o reprezintă in")











, dizaharide (30)% şi monozaharide. În alimentaţie glucidele.")
, dizaharide (30)% şi monozaharide. În alimentaţie glucidele.")

 consta in efectuarea unei diagrame familiale care cuprinde istoria si insusirile.")




 Starea solidă: au formă proprie; rigiditate; au volum propriu (incompresibilitate). b) Starea lichidă:")
 După tipul de hemoliză Streptococii beta-hemolitici (produce.")

")


