Скачать презентацию
1
Sindromul de malabsorbţie (curs studeni 2011) Confereniar Petru Martalog Catedra Pediatrie nr.1
 Confereniar Petru Martalog Catedra Pediatrie nr.1")
2
Prima datorie a unui medic constă în însuirea unei învăături serioase, reînnoite în permanenă. J. Bernard

3
Человек живет не тем, что он съедает, а тем, что переваривает. Положение это одинаково справедливо относится как к разуму, так и к телу (Бенджамин Франклин)
")
4
Sindromul de malabsorbţie (SM) – este un complex de manifestări clinice digestive şi extradigestive, datorat dereglări mecanismelor de digestie, absorbţie şi transportare ale principiilor alimentare.
 – este un complex de manifestări clinice digestive şi extradigestive, datorat dereglări mecanismelor de digestie, absorbţie şi transportare ale principiilor alimentare.")
5
Tubul digestiv pentru funcţia sa optimă necesită câteva condiţii: Intestinul să fie suficient de lung, traectoria digestivă să nu fie întreruptă sau deviată, să efectueze mişcări normale propulsive şi de amestec ale alimentelor cu secretele digestive; suprafaţa mucoasei să fie capabilă de a asigura digestia, absorbţia, transportul substanţelor nutritive; Fluxul secreţilor (gastrică, pancreatică, biliară, intestinală )să fie adecvat calitativ şi cantitativ; Circulaţia sangvină şi limfatică să asigure preluarea şi transportul substanţelor nutritive spre celulă. Tulburarea oricărei din aceste condiţii poate duce la apariţia fenomenului de malabsorbţie.

6
Digestia şi absorbţia sunt un complex de mecanisme de transformare şi transport ale principiilor alimentare către celulă. Ele se desfăşoară succesiv. Rolul esenţial în procesul de digestie – absorbţie îi revine enterocitului. Important: durata de viaţă medie a enterocitului este ore; fiecare 4-6 zile are loc înlocuirea totală a celulelor epiteliului, microvilelor...suprafaţa mucoasei intestinale de absorbţie la adulţi este circa 450 m² DIGESTIA – procesul de transformare fizico – chimică a principiilor alimentare în produşi asimilabili de către enterocit. Ea se realizează sub acţiunea sucului gastric, sucului şi enzimelor intestinale şi pancreatice, bilei (corespunde fazei intraluminale a digestiei) ABSORBŢIA – asigură transportul principiilor alimentare degradate din lumen în enterocit şi de aici în circulaţia sangvină v. porta sau limfatică. Transferul este activ, pasiv, prin difuziune, pinocitoză.

7
Fazele procesului de digestie–absorbţie intestinală Faza luminală (intraluminală) - este digestia alimentelor în lumenul intestinal sub acţiunea enzimelor pancreatice, acizilor biliari, enzimelor intestinale. Are loc degradarea alimentelor până la oligomeri (oligopeptide, oligozaharide etc. vezi tabela ). Faza membranară (parietală) – se petrece la nivelul marginii în perii a enterocitelor sub acţiunea enzimelor enterocitului. La acest nivel este o specificitate strictă (fiecare ingridient are enzimă specifică, mecanism de transport). Faza intracelulară – se produce intracelular, este în parte o nouă degradare, dar şi o resinteză (perioada neonatală).
 - este digestia alimentelor în lumenul intestinal sub acţiunea enzimelor pancreatice, acizilor biliari, enzimelor intestinale. Are loc degradarea alimentelor până la ol")
8
Produşi iniţialiProduşi ai digestiei intestinale (fazele) Transport în: Ingeraţi De prediges- tie Inraluminală (s.p; s.i; bila) Parietală (enterocit) Intacelulară (enterocit) Glucide (hidraţi de carbon) Polizahari- de (dextrane) Dizaharide, Oligozahari- de Monozahari- de Monozahari de (Glucoza, galactoza, fructoza). Sânge (vena portă) ProteinePolipeptide Peptone. Octopeptide Decopeptide Aminoacizi. Tetra – dipeptide. Aminoacizi. Aminoacizi Tri– dipeptide LipideTriglicerideMicele cu monogliceride Acizi graşi ChilomicroniTrigliceride Chilomicroni Limfa Tab.1 Etapele şi sediile procesului de digestie şi absorbţie
 Transport în: Ingeraţi De prediges- tie Inraluminală (s.p; s.i; bila) Parietală (enterocit) Intacelulară (enterocit) Glucide (hidraţi de carbon) Polizahari- de (dextrane) Dizaharide, Oligozaha")
9
Terminologie Digestia – absorbţia – transportul sunt etape succesive. Sindromul de maldigestie – dereglarea proceselor de digestie intaluminală prin insuficienţa pancreatică, biliară, intestinală. Sindromul de malabsorbţie –dereglarea proceselor de absorbţie şi transport al substanţelor deja digerate (celiachia). Dar separarea acestor 2 procese este convenţională, în literatură termen unic,, Sindromul de malabsorbţie.

10
Anomalii ale fazei intraluminale (digestie inadecvată) Anomalii ale fazei intestinale (boli ale mucoasei; absorbţia inadicvată) I. Dizordini (cauze) pancriatice Fibroza chistică Sdr. Schwachman Deficit izolat de lipază Pancreatita cronică II. Scăderea acizilor biliari Boli cronice hepatice (hepatita neonatală, ciroză) Atrezia biliară Colecistita cronică III. Dizordini (cauze) intestinale Contaminare bacteriană Sindromul intestinului scurt Hipo – şi aclorhidria Cauze anatomice cu stază intestinului subţire Sdr. imunodeficienţă Malnutriţia - marasmul Diareea intractabilă. I.Leziuni morfologice ale mucoasei Infecţii bacteriene, virale, micotice Infestaţii intestinale Intoleranţa la proteinele laptelui de vacă Boli inflamatorii cronice (Crohn,CUN) Boala celiacă Deficitul secundar de dizaharidaze Enteropatia autoimună Imunodeficienţe combinate, SIDA II.Cauze anatomice Boala Hirschprung Malrotaţii Stenoza jejunului Rezecţii intestinale III.Defecte primare de absorbţie Deficitul de dizaharidaze Malabsorbţia acidului folic Malabsorbţia primară Mg,Cu Malabsorbţia glucozei Abetalipoproteinemia Acrodermatitis enteropatică Deficitul de enterokinază Clasificarea fiziopatologică a sindromului de malabsorbţie
 Anomalii ale fazei intestinale (boli ale mucoasei; absorbţia inadicvată) I. Dizordini (cauze) pancriatice Fibroza chistică Sdr. Schwachman Deficit izolat de lipază Pancreatita cronică II. Scădere")
11
Defecte ale fazei de eliberare (transport) Boala Whipple Limfangiectazia intestinală Insuficienţa cardiacă congestivă Limfomul Cauze diverse Boli endocrine (diabet, hipoparatiroidism, Addison) Insuficienţa renală Dezordini imune Malabsorbţia poate fi selectivă, limitată doar la anumite nutriente (ex. malabsorbţia de glucide, de lipide, de vit.B 12, de magneziu). Malabsorbţia globală, generalizată care cuprinde mai multe elemente nutritive (glucide, lipide, proteine, microelemente).
 Boala Whipple Limfangiectazia intestinală Insuficienţa cardiacă congestivă Limfomul Cauze diverse Boli endocrine (diabet, hipoparatiroidism, Addison) Insuficienţa renală Dezordini imune Malabsorbţia poate fi")
12
Diareea cronică. Diareea cronică (DC) – eliminare zilnică de scaune modificate ca aspect, frecvenţă,fluiditate şi volum (peste 10g/kg/zi copil şi peste 300g/24ore la adult) cu o durat[ de peste 2 (3) săptămâni. Se consideră mai semnificative aspectul şi fluiditatea (eliminarea de apă scaunelor). Diareea cronică nu este sinonimă cu sindromul de malabsorbţie. Există diarei cronice cu şi fără sindrom de malabsorbţie. Exemplu: sindromul de colon iritabil cu diaree, dar fără malabsorbţie invers – malabsorbţia selectivă de Mg, Cu fără diaree evidentă. Delimitarea acestor 2 stări este dificilă, necesită observaţie clinică şi investigaţii. Mai frecvent diareea cronică este manifestare a sindromului de malabsorbţie.
 – eliminare zilnică de scaune modificate ca aspect, frecvenţă,fluiditate şi volum (peste 10g/kg/zi copil şi peste 300g/24ore la adult) cu o durat[ de peste 2 (3) săptămâni. Se consideră mai semnificative aspectul")
13
Cauze comune de diaree cronică. Copil mic Sindromul de malabsorbţie secundar (postinfecţios) Deficitul de dizaharidaze Fibroza cistică Intoleranţa la proteinele laptelui de vacă (soia) Dizordini alimentare Copil mare Diareea cronică nespecifică Deficitul de dizaharidaze (secundar) Malabsorbţia secundară (postinfecţioase) Celiachia Fibroza cistică Giardiaza Adolescent Sindromul intestinului iritabil Giardiaza Intoleranţa la lactoză Boli inflamatorii intestinale cronice (CUN,Crohn). Cauzele diareei cronice. Actualmente recomandă: Diarei severe prelungite DSP, intratabile diarrhea - la care nu se poate găsi o cauză precisă; Enteropatii cronice specifice – de etiologie clară.
 Deficitul de dizaharidaze Fibroza cistică Intoleranţa la proteinele laptelui de vacă (soia) Dizordini alimentare Copil mare Diareea cronică nespecifică Defic")
15
SimptomFiziopatologia Diareea cronică Deriglarea absorbţiei de apă şi elictroliţi Secreţia crescută de apă şi elictroliţi Acizi biliari dihidroxilaţi neabsorbiţi cu reducerea absorbţiei apei şi elictroliţilor Tranzit intestinal crescut. Caracteristica scaunilor (aspectul, culoarea, frecvenţa, volumul, mirosul, caracterul, prezenţa resturi patologice). Polifecalie, fetide Scaune steatoreice: polifecalie (voluminoase), fetide, grăsoase, lipicioase, alcaline, păstoase în celiachie, mucoviscidoză, boli pancreas Scaune apoase – lichide, miros acid, explozive, spumoase, eliminare frecvent în timpul alăptării – sugerează malabsorbţia glucidelor Corelaţie diaree – alimetaţie (IPLV, la zaharoză) Semne şi simptome de prezentare ale SM Etapa anamnestico – clinică care va preciza vârsta de debut al maladiei, semnele clinice şi evoluţia lor, semnele clinice digestive şi extradigestive; caracteristica şi durata diareei, cronologia apariţiei semnelor clinice, legătura diaree – alimentaţie, factorii ereditari.

16
Flatulenţă, Meteorism Dureri abdominale Anorexie Bulemie Distonie abdominală Fermentaţia glucidelor nedigerate de flora bacteriană colonică Distensia intestinală. Colici intestinale Inflamaţia intestinală În malabsorbţie În insuficienţă pancreatică Hipomotilitate, dismicrobism, hipotonie,acumulare de gaze Malnutriţie generalizată, persistentă,rezistentă la tratament Maldicestie Malabsorbţie generalizată Perderi de calorii Diaree cronică Anorexie, toleranţă digestivă scăzută Carenţă de minerale, vitamine, imunitar Dereglări trofice Paliditate, stomatită, glosită, hiperkiratoză Absorbţie redusă şi defecit de Fe, folaţi, vit A, B12, zinc ect. Hipoproteinemie Tetanie, parestezii Malabsorbţie de calciu, magneziu Deficit de cobalamină Dureri osoase Hipovitaminoză D, hipocalcemie, osteoporoză Retard psihomotor, insuficienţă endocrină, retard sexual Semne şi simptome de prezentare ale SM

17
Manifestări paraclinice în SM. Examinul coprologic macroscopic şi microscopic. Proba de digestie relevă prezenţa fibrelor musculare nedegerate (creatoree), amidon (amiloree),grăsimi (steatoree) trigliceride, grăsimi neutre din insuficienţă pancriatică exocrină. Lipidele în scaun nu trebuie să depăşească 4 -4,5g/24 ore. Prezenţa de acizi graşi, săpunuri, bilirubină netransformată – malabsorbţie intestenală. Cantitate mare de celuloză, bilirubină, floră iodofilă abundentă – tranzit intestinal accelerat. pH–ul acid – intoleranţă la lactoză. Prezenţa sîngelui în scaun (în lipsa infecţiei,giardiei) –IPLV. Prezenţa de mucus, leucocite, eritrocite – marcheri de afectare preponderent colitică (mucus se produce numai colon, hematiile se distrug în intestinul subţire)
, amidon (amiloree),grăsimi (steatoree) trigliceride, grăsimi neutre din insuficienţă pancriatic")
18
Determinarea grasimilor în masele fecale; Determinarea enzimelor pancreatice în sucul duodenal; Determinarea tripsinei în masele fecale Testul D-xiloza pentru studiul absorbiţiei intestinale; Xiloza este o hidrocarbură care se absoarbe pasiv la nivelul jejunului. După absorbţie se elumină prin urină peste 20% din cantitatea administrată. Se administrează oral 0,5 g/kg. Eliminarea cu urina sub % indică la lezarea mucoasei intestinale ( celiachie). Testele de încărcare şi provocare (cu lactoză, zaharoză, etc.); Biopsia intestinală; Determinări biochimice pentru carenţele digestive cronice: Anemie microcitară (carenţă de fier), macrocitară (carenţă de acid folic, B12); Hipocalcemie,hipomagnemie; Hipoproteinemie, hipoalbuminemie; Hipolipidemie; Reducerea factorilor de coagulare vit.K dependenţi; Examene radiologice, microbiologice.

19
CaracteristiciAlimentaţie naturală Alimentaţie artificială Copil mareScaune patologice ConsistenţaMoale – legat, Granular - moale Tare – format, granulat Cilindrice, tare. Lichide- apoase, spumoase, Păstoase – grăsoase. CuloareaGalben – aurie, la aer - verde Alb – gălbui, cenuşiu, galben Brună - galben Albă, verde, întunecată, sînge, mucus Frecvenţa3 – 6 pe zi1 – 2 pe zi1 pe zi1 – 2 pe zi MirosulPuternic acid, acrişor Fecal - dezagreabil Miros fecal, caracte- ristic Fetid, de putrefacţie AciditateaAcid, pH 4-6Alcalin, pH Alcalin neutru - Volum g/zi Polifecalie, peste 300g/zi Scaunele normale şi patologice la copii Examenul macroscopic al scaunelor

21
BOALA CELIACĂ Confereniar Petru Martalog

22
Ce este boala celiacă? Boala celiacă (BC) – este o afecţiune inflamator-imună a intestinului subţire, determinată de hipersensibilitate la gluten, survine la persoanele genetic predispuse, caracterizată clinic prin sindrom de malabsorbţie globală, histologic atrofie vilozitară jejunală, evolutiv - remisie clinico-histologică după excluderea glutenului din alimentaţie şi recădere histologică la reîntroducerea acestuia (proba reutei). Boala celiacă - cifrul K 90.0 Boala celiacă- coeliakia, din grec. Koilikos, suferină intestinală (sinonime celiachia, enteropatia indusă de gluten, intoleranţa la gluten, sprue tropical).
 – este o afecţiune inflamator-imună a intestinului subţire, determinată de hipersensibilitate la gluten, survine la persoanele genetic predispuse, caracterizată clinic prin sindrom de malabsorbţie globală, hi")
23
Epidemiologie. BC are o incidenţă variabilă, în funcţie de zona geografică. Incidenţa bolii clinic simptomatică variază în Europa de la 1/250 la 1/7000, în Franţa fiind de 1/2500. Conform studiilor epidemiologice recente, prevalenţa maladiei este în medie 1:300 (între 1:100 şi 1:500 în Europa şi SUA). BOALA CELIACĂ NU ESTE RARĂ…

24
Ţara Forme clinice evidente Prevalenţa screening Referinţe Finlanda 1:1000 1:99Malc, 2002 Suedia 1:330 1:177CarlssonA.K., 2001 N.Zelandă - 1:90Cook H.B.,2000 Norvegia - 1:340Hovdenac, 1999 Franţa 1:2000 1:388Butron-RuaultM,2002 UK - 1:122Johnston, 1998 Spania 1:1420 1:389Riestra S., 2000 Italia 1: :180Catassi,Fabiani, 2002 SUA 1: :133FasanoA., 2003 Russia - 1:1000 Sahara - 1:18Catassi, 2001 Brazilia - 1:50 Gandolfi L., 2000 Prevalenţa geografică a bolii celiace

25
Rezultatele sunt atribuite utilizării testelor serologice de screening (titrul de anticorpi antigliadină, antiendomisiali). BC are o distribuţie de vîrstă diferită. Perioade maxime de afectare sunt vîrsta pînă la 5 ani, apoi decada a 4-a a 5-a de viaţă. Raport F:B=2:1. Majoritatea copiilor cu BC prezintă forme atipice sau latente, frecvent nediagnosticate (contextul alte boli intestinale). Boala este rară în America de Nord, foarte rară în America Latină, în China i India, este excepţională în Africa neagră.
. BC are o distribuţie de vîrstă diferită. Perioade maxime de afectare sunt vîrsta pînă la 5 ani, apoi decada a 4-a a 5-a de via")
26
FACTORI DETERMINANI MAJORI ÎN BOALA CELIACĂ BC apare în rezultatul interacţiunii a trei factori obligatori: factorul exogen (de mediu) declanator factori genetici (ereditari) factori imunologici.
 declanator factori genetici (ereditari) factori imunologici.")
27
PATOGENIA BOLII CELIACE Factori genetici HLA Factori de mediu Gluten Răspuns imun Inflamaie BOALA CELIACĂ

28
Factorul exogen. Este glutenul –proteina din grîu, orz, secară, ovăs. Fracţia toxică a glutenului din grîu este gliadina. Gliadina (grupul prolaminelor, proteine de rezervă specifice gramineelor) are masa moleculară mare, conţinut înalt de glutamină, prolină. Similare gliadinei sunt prolaminele din orz (hordelina), secară (secalina), ovăs (avenina).
 are masa moleculară mare, conţinut înalt de glutamină,")
29
FACTORI DIETETICI GLUTENUL – factor responsabil Familia Graminee Subfamilie Grup ZizaneaeOrizeaeHordeaeAveneaeFestuleaeChlorideae Fesincoideae Secara Orz Orez salbatic OrezOvazMeiTeft Grau

30
Factori dietetici Factori trigger de mediu amidon 75% GRÂU 15% proteine GLUTEN GLIADINA (solubilă în alcool - prolamine) GLUTENINA (insolubila in alcool) Prolamina-grîu α gliadina Prolamina secară secalina Prolamina orz hordeina Prolamina ovăz avenina
 GLUTENINA (insolubila in alcool) Prolamina-grîu α gliadina Prolamina secară secalina Prolamina orz hordeina Prolamina ovăz avenin")
31
Proteinele cerealelor care pot provoca inflamaie intestinală în boala celiacă CerealeProlamineCompoziieToxicitate Grîu α - gliadina 63%G, %P +++ OrzHordeina63%G, %P ++ SecarăSecalina63%G, %P ++ OvăzAvenina% crescut G, % scazut P +-+- PorumbZeina% scazut G, % crescut P - Orez?-

32
Factori non-dietetici. Infecii virale: Secvene homologe între α gliadină i - Adevovirus tip 12 si 17 - Virus rubeolic - Herpes virus uman Infectii parazitare: Secvene omologe între α gliadină i - Plasmodium Yoelli Altele

33
Nu este atât de importantă vârsta la care se introduce făina de grâu în alimentaţie, ci mai ales cât timp este alimentat sugarul la sân, laptele de mamă fiind un real protector. Alăptarea la sân întârzie debutul bolii şi aceasta cu cât alăptarea este mai prelungită, independent de momentul introducerii făinii !!!

34
FACTORI GENETICI Riscul de boală celiacă – genetic determinat; Transmisie autozomal dominantă cu penetrană incompletă; Prevalena 10-15% la rude de gradul I; Predispozitie familiala puternica; Concordana la gemeni monozigoi 80%, frai 10%, copii 5-10%; Concordana de 30-40% la frai HLA identici sugerează alte gene implicate

35
GENETICA Asociere puternică cu antigene HLA clasa II DQ2/DQ8 localizate pe cromozomul 6p21 În Europa: HLA-DQ2 = 95% bolnavi HLA-DQ8 = 5% bolnavi Bolnavi care nu exprimă nici DQ 2 şi nici DQ 8 sunt excepţionali. În populaia generală cu stare de sănătate bună: - DQ2 = 25-30% - DQ8 = 5-15%

37
PATOGENIA BOLII CELIACE Teoria enzimatică. Explică BC prin insuficienţa de peptidază enterocitară, responsabilă de scindarea prolaminelor. Gliadina ar exercita efect toxic direct asupra mucoasei, cît i prin mecanismele imunologice sensibilizate de o predispunere genetică. Absenţa peptidazei – glutenul nu poate fi hidrolizat. Are loc acumularea de peptide toxice în mucoasă, preponderent jejunul inferior- ileonul. Are loc distrucţia accelerată a enterocitelor sub efectul glutenului. Compensator -proliferarea celulara, hipertrofia criptelor, ulterior –atrofie. Daca se respectă dieta, procesul se stopează. Efectul toxic al gliadinei a fost demonstrat i in cultura de organ (experiment).

38
Teoria patogenetică imunologică. Cea mai acceptată la ora actuală. BC este o consecinţă a hipersensibilizării mucoasei intestinale la gluten. Mecanismele imune sunt mediate celular şi umoral. Oligopeptidele imunogenice ale gliadinei sunt captate de celulele prezentatoare de antigen: macrofagele din lamina propria sau enterocitele (exprimă molecule HLA clasa II DQ pe suprafaţa lor), cu prezentare ulterioară limfocitelor T CD4+. Celule CD4 + din lamina propria, activate, vor declanşa răspunsul imun patologic.

39
Consecinele fiziopatologice. chemotactism cu hipercelularitate locală (mastocite, macrofage, eozinofile, bazofile, neutrofile); creşterea populaţiei limfocite intraepiteliale CD8+ cu rol citotoxic creşterea numărului de plasmocite cu stimularea producerii de anticorpi specifici; eliberare de citokine proinflamatori Th-1, IL-2, IL-6, TNF, IFN-gamma, IL-15 (chemokină), care exercită efecte citotoxice asupra enterocitelor şi favorizează proliferarea epitelială. leziuni intestinale specifice.
; creşterea populaţiei limfocite intraepiteliale CD8+ cu rol citotoxic creşterea numărului de plasmocite cu stimularea produ")
40
Transglutaminaza tisulară (tTG) Autoantigenul principal, ţintă a autoanticorpilor caracteristici ai BC Enzima de transamidare Ca-dependenta Modifica gliadina prin de-amidarea glutaminei Prezenta la toata tipurile de celule Este mecanismul autoimun al BC, rezultat al produciei de autoanticorpi anti tTG, anti EMA, anti AGA, anti RA.
 Autoantigenul principal, ţintă a autoanticorpilor caracteristici ai BC Enzima de transamidare Ca-dependenta Modifica gliadina prin de-amidarea glutaminei Prezenta la toata tipurile de celule Este mecanismul autoimun al")
41
Fiziopatologie. Distrugere accelerată a enterocitelor sub acţiunea glutenului, mecanismelor imune. Compensator se produce o mitozăa intensă dar insuficientă. Diminuarea suprafeţei de absorbţie intestinală cu sindrom de malabsorbţie generalizată. Insuficienţa dizaharidazelor i altor enzime locale duce la maldigestie. Perturbarea transportului grăsimilor, malabsorbţia hidrocarbonatelor prin transport redus. Diaree cronică-steatoree, malnutriţia, deficit de microelemente, vitamine, stresul metabolic i deficitul imunitar. Deficit de Ca, P, Mg, vit.K duc la osteoporoză. Nivele scăzute de hormon somatotrop cu retard statural.

42
Clasificare. Nu există clasificare de consens. Recomandă. BC simptomatică, circa 1/ /2000 BC silenţioasă, asimptomatică, circa 1/300 BC latentă Perioada bolii: activă, remisiune. Nu se cunosc cu precizie factorii care determină o formă sau alta a bolii şi trecerea din una în cealaltă. Totui: durata alimentaiei naturale, vîrstă cînd sa întrodus glutenul, i cantitatea de gluten în alimentaie sunt trei factori care determină cînd şi cum apare boala celiacă.

43
Care sunt manifestările clinice de BC? BC poate debuta la orice vârstă. Cel mai frecvent debut este între 8-24 luni, la circa 4-8 săptămâni de la introducerea produselor de gluten (grâu, secară, orz). Semnele BC cuprind manifestări digestive, extradigestive, sindromul pluricarenţial. Semne clinice: Gastrointestinale (clasice) Non gastrointestinale (atipice) Asimptomatice. Boala celiacă poate fi asociată cu alte boli în special: Boli autoimune Alte sindroame
. Semnele BC cuprind manifestări digestive, extradi")
44
Tablou clinic. Copil sub 2 ani Diareea cronică sau recurentă, steatoree Încetinirea creşterii sau pierdere în greutate Distensie abdominală cu abdomen mare Membre subţiri, lungi (copil păiangen) Anorexie, vărsături, dureri abdominale Deficit psihomotor, iritabilitate Constipaie (rar, la doar 15%)
 Anorexie, vărsături, dureri abdominale Deficit psihomot")
45
Semnele digestive tipice. Diareea cronică. Scaune repetate, de la 2-8 pe zi, mai frecvent 3-4 ori zi. Scaunele treptat devin voluminoase (polifecalie); consistenţa păstoasă, semilichidă (balegă de vacă); stetoreice, grăsoase, lucioase, lipicioase (se spală greu), miros fetid, deschise, acolice la culoare; prezenţa de resturi alimentare nedigerate, uneori diaree apoasă, după lapte (există deficit de lactază secundar); La 10-15% cazuri poate fi constipaţie. Se asociază o distensie abdominală (meteorism+lichid în intestin) - pseudoascită, încetinirea creşterii. Sunt caracteristice anorexia rebelă, rar vome. Treptat se stabileşte o malnutriţie staturoponderală severă cu dispariţia totală a ţesutului adipos, hipotonie, distrofie musculară, exterior caracteristic: copil trist, apatic, indiferent, ţesut adipos absent, membre subţiri şi lungi, abdomen voluminos (aspect de păianjen). Pielea subţire, se văd ansele intestinale.
; consistenţa păstoasă, semilichidă (balegă de vacă); stetoreice, grăsoase, lucioase, lipicioase (se")
46
SEMNE TIPICE DE BC

47
Manifestări extradigestive. Sindrom pluricarenţial: anemie prin carenţă în fier şi acid folic, rahitism, osteoporoză, dereglări trofice cutaneo- mucoase, tulburări psihice, deficit de dezvoltare somatică, deficit de dezvoltare psihomotorie, tulburări endocrine (pubertate întârziată, sterilitate), manifestări articulare (artrite), hemoragice (deficit de vitamină K). Crizele celiace- apar la asocierea deficitului dizaharidazic: diareea cu scaune frecvente, acide, păstos-apoase, vome, colici abdominale, prolaps rectal, deshidratare.

48
Boala celiacă atipică -se consideră că se întâlneşte la fiecare al patrulea copil cu BC. Cu cât copilul este mai mare, cu atât mai frecvent sunt forme atipice de BC. Pentru copilul mare şi adolescent: disconfort abdominal, inapetenţă, statură mică, anemie persistentă, osteoporoză prematură, dureri osoase, oboseală, slăbiciuni, diaree sau constipaţie. Manifestări osteomusculare: hipotrofie-distrofie, hipotonie musculară cu oboseală, dureri osoase, osteomalacie şi osteoporoză, hipoplazie dentară. Manifestări hematologice: epistaxis, gingivoragii, echimoze - prin deficit de vitamina K şi factori de coagulare. Manifestări mucocutanate: pielea uscată, palidă, turgor flasc, hipercheratoză, stomatite, dermatită herpetiformă, schimbări la nivelul unghiilor, părului. Manifestări neurologice: neuropatii periferice, encefalopatie dismetabolică. Manifestări endocrine: nanism, infantilism, hipotonie.

49
Manifestări clinice nongastrointestinale, atipice Cea mai comună formă de prezentare a BC la copil mare pînă la adult: Dermatita herpetiformă Hipoplazia smalului dentar – dini permaneni Osteopenie/ osteoporoză Artrita Dureri articulare i/sau osoase Statură mică Pubertate intarziată, sterilitate Anemia feriprivă inexplicabilă, rezistentă la fier oral Hepatita Epilepsie cu calcificări occipitale Oboseală cronică

50
DERMATITA HERPETIFORMĂ - maculă eritematoasă > papule urticariene > vezicule tensive - prurit sever - distribuie simetrică - 90% nu au simptome GI - 75% atrofie vilozitară - sensibilitate la gluten Garloch J.J. AT AL: Br. J. Dermatol.1994:131, 822 – 6. Fry L. Balliers: Clin Gastroenterol

51
DEFECTE SMAL DENTAR

52
OSTEOPOROZĂ

53
Ce alte probleme de sănătate prezintă pacienii cu boală celiacă? Boala celiacă este asociată cu alte boli autoimune: - Diabet zaharat tip I; - Tiroidită autoimună; - Boli ale esutului conjunctiv (LES, ACJ, SS); - Cardiomiopatie idiopatică; - Boala Addison - Dermatită herpetiformă - Alopeie - Hepatita autoimună
; - Cardiomiopatie idiopatică; - Boala Ad")
54
Această heterogenitate evolutivă a bolii şi descoperirea relativ recentă, datorită testelor diagnostice serologice, a prevalenţei crescute, în populaţia generală, a formei sale silenţioase caracterizată printr- o atrofie vilozitară asimptomatică, l-a determinat pe Ferguson să regrupeze aceste forme în iceberg-ul celiac în care, boala patentă nu este decât partea vizibilă

55
Iceberg-ul bolii celiace BC simptomatică 1/ /2000 BC silenţioasă 1/300 BC latentă

56
ICEBERG CELIAC

57
Diagnosticul diferenial. Sindromul colon iritabil Anemie feriprivă Boală inflamatorie intestinală cronică Diverticulită Sindromul de oboseală cronică Fibroza chistică de pancreas Insuficienţa dizaharidazelor Infestaţii parazitare- giardiaza (lambliaza) Anomalii intestinale Enterite infecţioase trenante

58
DIAGNOSTIC CLINIC SEROLOGIC HISTOLOGIC PROBA DIETETICĂ

59
Examenul coprologic: resturi nedigerate, fibre musculare, amidon, grăsimi - acizi graşi, saponine, celuloză, floră iodofilă. Testul cu D-xiloză pozitiv.

60
ENDOSCOPIE I BIOPSIE INTESTINALĂ

61
Biopsie jejunală. Leziunile histologice specifice (standardul de aur pentru diagnosticul boli celiace) ale mucoasei intestinului subţire sunt: Aplatisarea, turtirea suprafeţei mucoasei cu microvili scurtaţi Atrofia vilozitară totală sau subtotală (aspect în platou) Criptele intestinale sunt alungite (hiperplazia lor) Creşterea numărului de lomfocite intraepiteliale (IEL) în lamina propria (frecvent unica constatare) Infiltrat dens cu celule inflamatorii la nivelul laminei propria Scăderea raportului dintre aria epiteliului de suprafaţă şi epiteliul criptelor;
 ale mucoasei intestinului subţire sunt: Aplatisarea, turtirea suprafeţei mucoasei cu microvili scurtaţi Atrofia vilozitară totală sau subtotală (as")
62
Aspect macroscopic al mucoasei intestinale Boală celiacă Intestin normal

63
Atrofie intestinală parţială - aplatizarea vilozităţilor cu exocitoză la nivelul epiteliului de suprafaţă

64
Aspect microscopic al mucoasei intestinale Boală celiacă Intestin normal

65
Atrofie intestinală totală

66
TESTELE SEROLOGICE Anticorpi antigliadină (AGA) Anticorpi antiendomisium (EMA) Anticorpi antitransglutaminaza tisulară (anti-tTG) Tipare HLA
 Anticorpi antiendomisium (EMA) Anticorpi antitransglutaminaza tisulară (anti-tTG) Tipare HLA")
67
TESTE LE SEROLOGICE Identificarea indivizilor simptomatici care necesită biopsie Screening indivizilor asimptomatici cu risc Dovezi clare pentru diagnostic precoce Monitorizarea complianei dietetice (follow-up-ul BC non-invaziv) Înainte de testare pacienii trebuie să continuie alimentaia cu gluten Testele serologice sunt informative doar în faza activă a bolii La 1 lună de dietă fără gluten testele se negativează
 Înainte de testare pa")
68
ANTICORPI ANTIGLIADINĂ Anticorpi (clasa IgG i IgA) la proteina din gluten – grîau, orz i secară. Avantaje: Relativ ieftin i uor de lucrat Dezavantaje: Sensibilitate i specificitate scăzută.
 la proteina din gluten – grîau, orz i secară. Avantaje: Relativ ieftin i uor de lucrat Dezavantaje: Sensibilitate i specificitate scăzută.")
69
ANTICORPI ANTIENDOMISIUM - EMA Anticorpi tip IgA i IgG împotriva reticulinei esutului conjunctiv din jurul fibrei musculare netede. Avantaje: Sensibilitate i specificitate inaltă Dezavantaje: Fals negativ la copil mic Dependent de operator Scump i consumator de timp Fals negativ în deficitul de IgA

70
TRANSGLUTAMINAZA TISULARĂ - TTG Anticorpi tip IgA împotriva transglutaminazei tisulare Avantaje: Sensibilitate i specificitate crescută (TTG umană) Nu depinde de operator (ELISA/ RIA) Relativ ieftin Dezavantaje: Fals negativ la copilul mic Fals negativ în deficit de IgA Posibil mai puin specific ca EMA
 Nu depinde de operator (ELISA/ RIA) Relativ ieftin Dezavantaje: Fals negativ la copilul mic Fals negat")
71
TESTE SEROLOGICE COMPARATIV Sensibilitate Specificitate %______________________________________________________ AGA – IgG 69 – 85 % 73 – 92% AGA – IgA 52 – 90 % 82 – 94% EMA (IgA) 85 – 98 % 98 – 100% TTG (IgA) 90 – 98 % 94 – 96% Testele serologice au sensibilitatea şi specificitatea înaltă (cca %) când se cuplează AGA+EMA (sau AcTG), izolat mai puţin. Testul serologic cel mai fiabil rămâne dozarea anticorpilor anti-endomisium Dozarea anticorpilor simplifică procedura diagnostică şi urmăreşte evoluţia bolii celiace, dar nu poate substitui biopsia de mucoasă jejunală.______________________________________________________________ FARELL R aud KELLY CP.Am.J.Gastroenterol 2001:96:3237–46
 85 – 98 % 98 – 100% TTG (IgA) 90 – 98 % 94 – 96% Testele serologice au")
72
TESTE HLA Alele HLA asociate cu boala celiacă DQ2 – 95% bolnavi celiaci DQ8 – 15% bolnavi celiaci DQ2 – 30% populatie generala Valoarea testarii HLA. Valoare predictivă puternic negativă Negativitatea DQ2/DQ8 - exclude diagnosticul de B.C. 99% Schuppan Gastroenterology 2000: 119: 234 Kaukinen Am.J.Gastroenterol.2002, 97:695.

73
Examene hematologice: anemie microcitară (rar macrocitară – deficit vit. B 12 ), hipoproteinemie, hipolipidemie, hipoglicemie, hipocalcemie, hipofosforemie, hipomagnezemie, hipokaliemie, hiposideremie, poilhipovitaminoză. Ecografia organelor interne. Examen coprocitologic, coprocultura. Examenul radiologic baritat al tractului digestiv: dilatarea anselor cu lichid în ele, gaze. Osteoporoza oaselor, vârsta osoasă întârziată.
, hipoproteinemie, hipolipidemie, hipoglicemie, hipocalcemie, hipofosforemie, hipomagnezemie, hipokaliemie, hiposideremie, poilhipovitaminoză. Ecografia organelor interne.")
74
Aceste date au determinat Societatea Europeană de Gastroenterologie Pediatrică (ESPGHAN) de a revizui criteriile de diagnostic ale bolii celiace: Biopsia mucoasei intestinale cu atrofie vilozitară, cripte adânci, IEL, infiltrat limfoplasmocitar în corion Remisiune netă sub regim fără gluten, cu reluarea curbei ponderale ascendente (săptămâni până la 4-6 luni); remisia histologică în 6-12 luni; Testul de provocare (contra-proba proba de reşută) prin reintroducerea glutenului (nu este obligatorie dacă copilul are peste 2 ani); AAG, AEMA, şi anti-tTG dacă sunt net pozitivi în faza iniţială şi se negativează sub regim de excludere sunt argumente foarte serioase de confirmare a diagnosticului de boală celiacă.
 de a revizui criteriile de diagnostic ale bolii celiace: Biopsia mucoasei intestinale cu atrofie vilozitară, cripte adânci, IEL, infiltrat limfoplasmocitar în co")
76
Cum este tratată Boala Celiacă Singurul (unicul) tratament pentru BC de peste 50 ani este dieta gluten – free. Regimul igieno-dietetic constă în excluderea totală a glutenului din alimentaţie, lucru aparent simplu. Complexitatea tratamentului constă în: Respectarea categorică a regimului fără gluten toată viaţa Recunoaşterea şi excluderea tuturor formelor de alimente ce conţin gluten Asigurarea unei balanţe echilibrate în principii nutritive (proteine, lipide, glucide, calorii) pentru creştere şi dezvoltare, calitatea vieii.
 tratament pentru BC de peste 50 ani este dieta gluten – free. Regimul igieno-dietetic constă în excluderea totală a glutenului din alimentaţie, lucru aparent simplu. Complexitatea tratamentului constă")
77
Cîteva reguli de conduită terapeutică a BC. 1. Evitarea categorică a alimentelor ce conţin gluten: grîu, secară, orz pentru toată viaţa 2. Evitarea în prima fază a ovăsului, ulterior fulgii de ovăs pot fi folosiţi 3. Folosirea din cereale numai a orezului, hrişca, porumb, soia, făina din topoică, ovăs 4. Monitorizarea tratamentului în clinică specializată prin determinarea anticorpilor specifici 5. Asigurarea consultării regulate a ununi medic nutriţionist 6. Citirea atentă a tuturor etichetelor la produsele alimentare şi la ingredientele folosite, prudenţă la aditivi alimentari, prudenţă la medicamente ce pot conţine gluten. 7. Instruirea intensivă şi extensivă a familiei şi a pacienţilor de către medic şi nutriţionist. 8. La etapa iniţială se exclude din alimentaţie şi lactoza. 9. Repetarea biopsiei intestinale dacă succesul clinic nu este corespunzător.

78
Surse de gluten sigure, evidente Cereale: Grau, Secara, Orz, ovaz Pâine, Cakes, Piscot, prajitura paste/taitei, crupe gri, arpaca, făina Patiserii, placinte/budinci Surse de gluten ascunse Excluderea salamurilor fierte, crenvurştilor; semifabricate din carne şi peşte; conserve din carne, peşte, legume, fructe, pastă de tomate, ketchup, îngheţată, iaurt, cşcaval, bastonae crabi, margarină, maioneză, sosuri pentru salate, bulion/supe în cuburi, unele tipuri de ceai, cafea, cacao, cvasul, vodca, berea, whisky. Aditivi alimentari ca amidonul, conservanii, stabilizatorii

79
Recomandări pentru sugari. Practic toate amestecurile lactate adaptate i cele curative nu conin gluten. Primele diversificări cu pireu din legume (cartofi, morcov) şi crupe fără gluten (orez, hrişcă, porumb). Firma Nestle are produse pentru treapta 3 fără gluten. Copilul după 12 luni Clinutren –Junior.
 şi crupe fără gluten (orez, hrişcă, porumb). Firma Nestle are produse pentru treapta 3")
81
ALIMENTE PERMISE carne, pete, ouă, lactate hrişcă, porumb, in, orez, sago, seminţe, sorg, soia, tapioca, boboase, cartofi, inclusiv faină din axeste produse legume i fructe, nuci, salam i crenvuti calitate superioară, ciocolată, marmeladă, unele caramele i îngheată, zefir Amidon din porumb, cartofi, orez Oet i alcool: oet distilat i spirt distilat sunt gluten-free

82
1. Regimul igieno-dietetic este unica condiţie pentru întreruperea mecanismelor patogenetice de BC. 2. Excluderea absolută a glutenului din făina de grâu şi a prolaminelor înrudite din secară, orz, (ovăs?) este unicul tratament eficient. 3. Regimul fără gluten (agliadinic) va trebui respectat cu stricteţe toată viaţa, indiferent de forma, gravitatea bolii, vârsta pacientului. 4. Regimul fără gluten nu comportă nici un efect advers asupra creşterii, dezvoltării copilului, calităţii vieţii, încadrării în societate etc.
 este unicul tratament eficient. 3. Regimul f")
83
Tratamentul medicamentos-are caracter adjuvant. Substituţie cu enzime pancreatice pentru procesul de digestie - atenţie la ambalaj să nu conţină gluten dismulat.Preparat de elecţie este Creon 10000, Corecia biocinozei intestinale- Bifiform, Dufalac. Tratamentul poluării bacteriene intestin: Enterofuril. Frecvent colestază, necesită hepatoprotectoare, coleretice de 2-3 ori/an, cure de 2-4 săpt. Tratamentul de substituţie: Fier, acid folic, calciu, vitamina A, D, K, E, selen, zinc Tratamentul malnutriiei după principiile generale.

84
Tratamentul crizei celiace- reechilibrare hidroelectrolitică şi acido-bazică, aminoacizi î n perfuzie. Tratament cu prednizolon- î n forme severe de BC. Complicaţii: Criza celiacă, malnutriţie severă, limfom intestinal-pînă la 15%, perforaţie sau stenoze intestinale, osteoporoză, convulsii. Supravagherea. Pentru toată viaa. Primii 2 ani de la stabilire diagnostic- trimstrial. Ulterior în condiii de evoluie bună- 1 dată pe an. Endoscopie i examene serologice- la 6-12 luni distană, în cazuri de recurene. Examene serologice- după posibilităi anual. Rudele pacientului- examene serologice, dacă este suspiciune este indicată endoscopia cu biopsie. Vaccinarea- în remisiune după scheme de cruare.

85
Ca i concluzii. Celiachia este boală genetică, persoanele nu pot tolera glutenul, o proteină din grâu, secară, orz. Netratată boala celiacă duce la atrofie intestin subire i malabsorbie intestinală globală. Fără tratament pacienii cu boala celiacă pot dezvolta complicatii ca osteoporoză, anemie, i cancer. Pacienii cu boala celiacă pot avea simptome de boală evidente, dar mai frecvent nu au semne clinice tipice. Diagnosticul implică teste serologice i, în cele mai multe cazuri, o biopsie a intestinului subire. Deoarece boala celiacă este ereditară, membrii familiei trebuie să fie testai. Boala celiacă are un singur tratament - eliminarea absolută a glutenului din dieta. Dieta fară gluten este pentru toată viaţa. Un nutriionist poate ajuta o persoana cu boala celiacă în selectarea produselor alimentare.

86
PÂÎNEA NOASTRĂ CEA SPRE FIINĂ... Evanghelia după Matei…

89
FIBROZA CHISTICĂ (MUCOVISCIDOZA) curs studeni 2011 Dr. confereniar Petru Martalog Catedra Pediatrie nr.1
 curs studeni 2011 Dr. confereniar Petru Martalog Catedra Pediatrie nr.1")
90
Ce este fibroza chistică (FC)? FIBROZA CHISTICĂ (FC) – este o boală ereditară monogenică sistemică cu afectarea glandelor exocrine (respirator, digestiv, reproductiv) care produc o secreie vîscoasă, săracă în sodiu i apă, cu obturarea canalelor excretorii (pancreatice, bronice etc.), evoluie gravă i prognostic nefavorabil. Unul din 2500 de copii se nate cu fibroză chistică Te-ai gîndit că ar putea fi copilul...?
? FIBROZA CHISTICĂ (FC) – este o boală ereditară monogenică sistemică cu afectarea glandelor exocrine (respirator, digestiv, reproductiv) care produc o secreie vîscoasă, săracă în sodiu i apă, cu obturarea canalelor excre")
91
FIBROZA CHISTICĂ transmitere autosomal- recesivă este cea mai frecventă boală monogenică a rasei albe frecvena variază după originea etnică: 1:3000–1:5000 nou-născui în Europa, America 1:17000 rasa neagră 1: >19000 asiatici. durata medie a vieii este de ani, deci FC nu mai este o afeciune pur pediatrică este o afeciune multisistemică a copilului i adultului caracterizată prin boală pulmonară cronică progresivă (obstrucie cronică i infecie cronică a căilor respiratorii) i maldigestie determinată de insuficienă pancreatică exocrină.

92
Частота встречаемости МВ в ряде стран* Страна Частота встречаемости Финляндия 1 : Турция 1 : Швеция 1 : Польша 1 : Россия 1 : Дания 1 : Испания 1 : Германия 1 : Чехия 1 : Соединенное королевство 1 : Италия 1 : Франция 1 : Швейцария 1 : Ирландия 1 : США 1 : Бразилия 1 : Куба 1 : Объединенные Арабские Эмираты 1 : 15,876 Индия 1 : – Япония 1 : – Австралия 1 : Примечание: * From Report of a Joint Meeting. The Molecular Genetic Epidemiology of Cystic Fibrosis // WHO / HGN / CF / WG / P. 15.

93
CF median survival, '85'86'87'88'89'90 '91'92'93 '94'95'96'97'98'99 '00 '01'02 '03 '04 year Median survival (years) '05 06'07 Net increase in UK 130 people/year
 '05 06'07 Net increase in UK 130 people/year")
94
Date de biologie moleculară. Bazele genice ale bolii. Gena patologică identificată pe cromozomul 7 – 7q3.1.3 (1989). Ea codifică sinteza unei proteine denumită CFTR (cystic fibrosis transmembrane conductance regulator) CFTR formată din 1480 aminoacizi ce aparine glicoproteinelor P Gena CF este genă mare de circa kilobaze, conine 27 exoni (zone de codificare) cu un ARN mesager~6,5 kilobaze. Descrise > 1500 mutaii, 20 mutaii frecvente Cea mai răspîndită mutaie în Europa este F 508 (delta F508 - absena fenilalaninei in poziia 508) - circa 70%.
. Ea codifică sinteza unei proteine denumită CFTR (cystic fibrosis transmembrane conductance regulator) CFTR formată din 1480 aminoaciz")
95
Dacă ambii părini sunt purtători ai unei gene defecte, atunci: ansa ca un copil să se nască cu fibroză chistică (mucoviscidoză) este de 25%; ansa ca un copil să fie doar purtător al genei defecte este de 50%; ansa ca un copil să nu mostenească gena defectă, adică nici să nu fie bolnav, nici să nu fie purtător al genei defecte este de 25%. Cei mai muli purtători ai genei defecte nu tiu că au această genă, deoarece ei sunt complet sănătoi. Majoritatea persoanelor află că sunt purtătoare ale acestei gene dacă: au un copil cu fibroză chistică (mucoviscidoză); au o rudă afectată de această boală; Dacă fac un screening prenatal (testează fătul în timpul sarcinii).
 este de 25%; ansa ca un copil să fie doar purtător al genei defecte este de 50%; ansa ca un copil să nu mostenească gena d")
97
Fotografia de mai sus ilustrează faptul că ambii părini sunt purtători ai genei defecte i că există o ansă de 25% ca un copil să se nască cu FC i 75% ca să se nască un copil sănătos. Dar, la fiecare copil conceput separat (excepie gemenii identici!) riscul este acelai de fiecare dată, iar ceea ce s-a întamplat la ultima sarcină nu crete sau scade riscul de avea un copil bolnav la sarcina viitoare. ansele de a avea un copil bolnav sunt aceleai la fiecare sarcină! Este imposibil de prevăzut ce se poate întampla la o anumită sarcină. Doi părini purtători pot avea mai muli copii bolnavi sau pot avea copii sanatoi i copii bolnavi. Doar testul genetic prenatal poate stabili dacă copilul incă nenăscut este sau nu afectat de fibroză chistică.
")
98
Bazele genice ale bolii. Gena FC codifică (reglează) sinteza unei proteine specifice denumite condiional CFTR (MVTR)- reglator transmembranar al fibrozei chistice (Cystic Fibrosis Transmemranare Conductance Regultor). CFTR- este localizată în zona apicală a celulelor epiteliale secretoare de mucus (vezi desen), situate la nivelul canaliculelor pancreatice, arborelui bronic, în intestin, glande sudoripare, caile biliare intrahepatice, glandele sexuale (masculine), mai puţin mucoasa nazala, glandele lacrimale, salivare. CFTR este un canal propriu-zis al ionilor de Cl, cît i reglează transportul electrolitic (Cl, Na, H 2 O) dintre aceste celule i lichidul interstiial.
 sinteza unei proteine specifice denumite condiional CFTR (MVTR)- reglator transmembranar al fibrozei chistice (Cystic Fibrosis Transmemranare Conductance Regultor). CFTR- este localizată în zona ap")
99
Bazele genice ale bolii. Principala funcie a proteinei CFTR este transportul (stimulat de cAMP) clorurilor spre i dinspre lumenul epitelial la nivelul membranei apicale a celulelor epiteliale. În mod fiziologic mişcarea apei în spaţiu intra- intercelular e determinată de transportul activ al ionilor de Cl, Na i apa îl urmează pasiv. Excepţie fac glandele sudoripare care au funcţie de absorbţie a Cl i nu de secreie! (test diagnostic) ANDERSON – prima descriere în 1938
 clorurilor spre i dinspre lumenul epitelial la nivelul membranei apicale a celulelor epiteliale. În mod fiziologic mişcarea apei în spaţiu intra- intercel")
100
Consecine. Lipsa proteinei CFTR (a canalului de Cl) determină blocarea, deficit de secreie activă a ionilor de Cl, urmată de incapacitatea celulelor de a secreta lichid (Na+H 2 O), cît i absorbţia crescută de apă (epiteliu respirator, intestinal). Secreiile celulelor exocrine vor fi anormale, vîscoase, lipicioase, deshidratare, greu de mobilizat (conţinut scăzut de Cl, Na, H 2 O, bogat în Ca, proteine). Are loc stagnarea, acumularea de mucus şi obstruarea canalelor excretorii pancreatice, bronhiilor mici, căilor biliare cu manifestări clinice corespunzătoare.
 determină blocarea, deficit de secreie activă a ionilor de Cl, urmată de incapacitatea celulelor de a secreta lichid (Na+H 2 O), cît i absorbţia crescută de apă (epiteliu respirator, intestinal). Se")
101
Celulă epitelială normală. Celulă epitelială în FC.

102
În fibroza chistică (mucoviscidoză) sunt afectate toate organele cu secreie internă.
 sunt afectate toate organele cu secreie internă.")
103
PATOGENIE Lipsa proteinei CFTR disfuncia transportului transepitelial al clorului Secreii mucoase cu coninut scăzut de apă Incapacitate de a elimina secreiile mucoase Coninut crescut în electrolii al sudorii i al altor secreii seroase Infecii cronice limitate la tractul respirator

104
ELEMENTE FIZIOPATOLOGICE ESENIALE în FC Gena patologica a FC pe cromozomul 7. Lipsa proteinei CFTR. Blocarea secretiei ionilor de Cl, Na, H 2 O la nevelul membranei apicale a celulelor secretoare de mucus. Eliminare de secret vîscos, abundent, deshidratat Obstrucţia canalelor excretorii, dereglarea drenajului glandelor exocrine Acumulare de mucus, dilataţia secundară canalelor excretoare a pancreasului ale bronhiilor

105
Staza de durată a secretelor Distensie pseudochistică a acinilor cu fibroză, insuficienă pancreatică exocrină. Dilatarea canaliculară a glandelor traheobroniale, stază, atelectazii, fibroză, broniectazii. Asocierea infeciei.

106
Consecinţe Stază - dilatare- hipertrofia ţesutului conjunctiv (fibroză) tusa reflectorie, la acumulare de mucus, frecvent noaptea, dispnee, accese de sufocare atrofie acinară, fibroză interstiţială -insuficienţă pancreatică exocrină staza cu leziuni obstructive: IRA, bronşiolite, bronşite obstructive, tusă cronică tip pertusis maldigestie (lipsă suc- enzime pancreatice, suc intestinal) IRA, pneumonii recurente, atelectazii, malabsorbtie secundară generalizată afectarea motilităţii ciliare, lipsa drenajului, bronşiectazii malnutriţie infecţie cronica asociată semne clinice emfizem pulmonar, fibroză pulmonară Boală pulmonară cronică progresivă cord pulmonar, deces
 tusa reflectorie, la acumulare de mucus, frecvent noaptea, dispnee, accese de sufocare atrofie acinară, fibroză interstiţială -insuficienţă pancreatică exocrină staza cu leziuni")
107
Fiziopatologia FC Anomalia genei CFTR Perturbarea transportului ionilor de clor Scaderea cantitatii de lichid de la suprafata CR Clearance mucociliar ineficient Obstrucie Infecie Inflamaie

108
MORFOPATOLOGIE. Afectarea fundamentală este modificarea calitativă i cantitativă a secreţiilor mucoase (vîscoase, aderente). Consecinţele acestor modificări sunt: obliterarea canalelor excretoare, dilataţia chistica ale glandelor, distrofia glandulară, fibroză chistică ale glandelor mucoase.
. Consecinţele acestor modificări sunt: obliterarea canalelor excretoare, dilataţia chistica ale glandelor, distrofia glandular")
109
MANIFESTARI CLINICE ÎN F.C. Clasificarea (este convenţională) Forma pulmonară 15-20% Forma intestinală - 5% Forma mixtă 75-80% Ileus meconial 10-12% Alte forme 1-4% La naştere copiii sunt aparent sănătoi. Boala debutează de obicei în copilărie: pînă la 6 luni - 65%, pînă la 12 luni la 80%, pînă la doi ani la 90%. Conform CIM (ICD 10), FC este în seciunea boli Endocrine, Nutriie i Metabolsm, Cap. 4. E84.0 CF with pulmonary manifestations, E84.1 CF with intestinal manifestations, E84.8 CF with other manifestations, E84.9 CF unspecified.
 Forma pulmonară 15-20% Forma intestinală - 5% Forma mixtă 75-80% Ileus meconial 10-12% Alte forme 1-4% La naştere copiii sunt aparent sănătoi. Boala debutează de obicei în copilărie: pînă")
110
Cînd suspectăm fibroza chistică? MANIFESTĂRI DIGESTIVE ÎN FC Ileus meconial al nou-născutului Icter neonatal prelunjit Insuficienţă pancreatică exocrină Diaree cronică Falimentul creterii staturoponderale Abdomenul dureros recurent Prolaps rectal Reflux gastro-esofagian Hipovitaminoze (A, D, E, K, B 12 ): xeroftalmie, tulburări trofice, rahitism, scăderea sintezei de PG, hemoliză,distrofie neuroaxonală, diateză hemoragică, anemie pernicioasă Pancreatită acută, recurentă, calcificări pancreatice. Colelitiaza şi colecistita Hipoproteinemia sugarului cu edeme, acrodermatită

111
Disease manifestations of CF with age

112
Manifestări clinice la sugar Diaree cronică Scaune steatoreice, neformate, voluminoase, fetide Icter neonatal prelungit Reinere în dezvoltarea fizică Prolaps rectal Tuse cronică, dispnee, wheezingul Bronite, bronsiolită, pneumoni recidivante Gustul sărat al pielii (transpiraie sărată) Crize de deshidratare la călduri mari Hipoelektrolitemie (Na, Cl) constantă Hipoproteinemie cu edeme Antecedente familiale de decese copii sugari, sau prezenţa de fraţi cu manifestări clinice similare

113
Manifestări clinice la precolari Tuse cronică, persistentă cu sau fără spută purulentă Dispnee cronică inexplicabilă, sindrom de oboseală cronică Simptom degete de toboar" Diaree cronică Retard staturo-ponderal sever Prolaps rectal Invaginaie intestinală Cristale de sare pe piele Crize de deshidratare hipotonice Hipoelektrolitemie (Na, Cl) constantă şi alcaloză metabolice Hepatomegalie sau dereglarea funcţiei hepatice inexplicabilă

114
Manifestări clinice la copil de vârstă şcolară Simptome respiratorii cronice de etiologie necunoscută Pseudomonas aeruginosa în spută Sinusită cronică Polipoză nazală Bronsiectazii pulmonare Simptom bastonae de toboar Diaree cronică Pancreatită Sindromul obstrucţiei intestinale distale Prolaps rectal Diabet zaharat în combinaţie cu simptome respiratorii Hepatomegalie. Boală cronică hepatică inexplicabilă Pubertate întârziată.

115
Manifestări clinice la adolescenţi şi adulţi Boală cronică pulmonară de etiologie necunoscută Bronsiectazii. Sinusită cronică. Polipoză nazală. Simptom bastonae de toboar Pancreatită. Sindrom de obstrucţie intestinală distală Diabet zaharat în combinaţie cu semne respiratorii Semne de ciroza hepatică si hipertensiune portala Retard statural Pubertate întârziată Sterilitate cu azoospermie la bărbaţi. Scăderea fertilităţii la femei.

116
MANIFESTĂRI DIGESTIVE Manifestari digestive: prezente la circa 80-90% din bolnavi, frecvent sunt punctul de plecare în stabilirea diagnosticului, se traduc prin insuficienţa pancreatică, ileus, malabsorbţie, afectări hepatobiliare. ILEUS MECONIAL, prezent la 12-15% din nn cu FC, prima manifestare a bolii. acumulare de meconiu vîscos bogat în proteine obstrucie ileon distal deshidratat

117
MANIFESTĂRI DIGESTIVE ILEUS MECONIAL. Ecografia fetală (al 2-lea trim.) –intestin hiperecogen Clinic: Lipsa meconiului in primele 48 ore Semne de ocluzie intestinală: vărsături cu bilă, distensie i meteorism abdominal, deshidratare, rar perforaţie-peritonită. Radiologic: dilatarea anselor intestinale, nivele hidroaerice in intestinul subire, absenţa aerului in colon, la nivelul ocluziei-fenomenul Sticlă mată. Echivalent de ileus meconial (Jensen 1962) = sindrom de obstrucie intestinală distală: dureri abdominale colicative obstrucie intestinală pariala sau completă prin materii fecale = formaiune palpabilă (mase fecale) - regiunea cecală sau colon ascendent, constipaie, invaginaie, volvulus
 –intestin hiperecogen Clinic: Lipsa meconiului in primele 48 ore Semne de ocluzie intestinală: vărsături cu bilă, distensie i meteorism abdominal, deshidratare, rar perforaţie-pe")
118
MANIFESTĂRI DIGESTIVE INSUFICIENA PANCREATICĂ EXOCRINĂ 85-90% = absena secreiei exocrine maldigestie Manifestarile sindromului de malabsorbţie: La sugarul mic semnul de alarmă este lipsa creşterii in greutate la copil care mănîncă bine, rar edeme hipoproteice, scaun instabil. La diversificarea alimentaţiei apare diareia cronică i falimentul creterii. Scaune steatoreice cu particule nedigerate, masele fecale devin voluminoase-polifecalie (4-5 ori mai mult faă de copil sănătos), devin pastoase-aspect grăsos, lucioase, fetide, aderente de scutec (oală lasă pete grase). Rar pot fi constipaţii, dar scaunele voluminoase, fetide, lipicioase, cu multe grasimi neutre. Se asociază malnutriţia staturo-ponderală severă, carenţa de vitamine si minerale, abdomen mărit în volum.

119
MANIFESTARI DIGESTIVE PROLAPS RECTAL - determinat de Volumul excesiv al scaunului Scăderea tonusului muscular datorită malnutriiei i creterii presiunii intraabdominale ANOMALII ALE TRANSPORTULUI LA NIVELUL MUCOASEI INTESTINALE deficit de absorbie a aminoacizilor malabsorbia sărurilor biliare

120
MANIFESTĂRI HEPATOBILIARE Colestaza sugarului Steatoza hepatică a copilului mic Ciroza biliară focală Ciroza multilobulară a adolescentului Colangita sclerozantă Litiaza biliară

121
MANIFESTĂRI HEPATOBILIARE MANIFESTĂRI HEPATOBILIARE Colestaza sugarului, - e rară În primul an de viaă disfuncia hepatică e adesea limitată la creterea transaminazelor Steatoza hepatică Hepatomegalie Ecografia hepatică i biopsia confirmă prezena lipidelor în ficat Ciroza biliară focală Expresie clinică absentă sau doar hepatosplenomegalie Cretere tranzitorie a transaminazelor i GT Ciroza multilobulară – la adolescent i adult Cînd testele hepatice se modifică e deja instalată Prima manifestare poate fi hemoragia digestivă

122
Manifestările respiratorii în FC. Sunt prezente la circa 90% din bolnavi Debutează precoce, determină evoluia bolii. Sunt consecinţa acumulării de mucus, atelectazii, incapacitatea mucociliară de a elimina mucusul i germenii patogeni. Tuse persistentă, cu spută în exces, expectoraţia dificilă a unui mucus vâscos, aderent Tusea apare precoce ca act reflector de acumulare de mucus, devine persistentă, frecvent nocturnă, uneori in accese chinuitoare (tip pertusis). Tusea provoacă oboseala cupilului.

123
Manifestările respiratorii în FC. Infecii respiratorii repetate -bronşite, bronşiolite, pneumonii Dispneea, accese de sufocare, sindromul bronho- obstructiv recurent – wheezingul. Atelectazii lobare sau segmentare Puseele infecioase sunt determinate de Staph. aureus, Haemofilus influenzae, Pseudomonas aeruginosa, Aspergillus fumigatus – se asociază cu evidenta progresare a leziunilor bronhopulmonare. Broniectazii, emfizem pulmonar, inflamaţie cronică Fibroză pulmonară, boală pulmonară cronică progresivă Cord pulmonar. Insuficienă respiratorie. Deces. Aspectul fizic al toracelui caracteristic: forma globuloasă, stern proeminent, spate cifotic.

125
ALTE MANIFESTĂRI CLINICE ÎN FC Pancreatita acută recurentă Sterilitate masculină: azospermia prin obliterarea canalelor care elimină sperma din testicule spre uretra. Funcia sexuală este normală, dar nu pot avea copii. Poate fi semn unic la bărbaţi. Crize de deshidratare prin pierderi de sodiu, apă, la călduri mari Transpiraie abundentă i foarte sărată- este observat de către părini atunci cînd sarută copilul sau apar cristale de sare ORL: Sinuzite purulente, Rinite cronice, Polipoză nazală Cardiace: Cardiomiopatie acută cu necroză miocardică, Cardiomiopatie dilatativă hipokinetică Osteoarticulare: osteoartropatia hipertrofică pulmonară şi artrite nespecifice Maligne: incidenţă mai crescută a malignităţilor

126
Diagnosticul fibrozei chistice EVALUAREA FUNCIEI PANCREATICE Examenul coprologic: scaune cu aspect lutos, steatoreic, cantitate mare de grăsimi (peste 4,5-5g/zi), prezenţa fibrelor musculare nedigerate, grăsimi neutre în cantitate mare, pH alcalin (insuficienţa pancreasului exocrin); Dozarea tripsinei în masele fecale: scădera sau lipsa ei; Dozarea albuminelor în meconiu: >20 mg/g de meconiu- test screening neonatal; Determinarea elastazei-1 fecale Determinarea enzimelor pancreatice în sucul duodenal; Ecografia abdominală: schimbări în pancreas, steatoză hepatică; Radiografie abdominală: bule de aer în ileus meconial.
, prezenţa fibrelor musculare nedigerate, grăsimi neutre în cantitate mare, pH alcalin (i")
127
Diagnosticul fibrozei chistice TESTUL SUDORII - rămîne standardul de aur în FC Testul sudorii la concentraţia de clor, care este influenţată de gradul transpiraţiei. Se poate obţine cu stimulare prin ionoforeză cu pilocarpină. Valorile normale sunt de 40 mmol/l. Testul este pozitiv cînd concentraţia de clor creşte >60mmol/l. Sunt necesare 3 teste la intervale peste o zi. Valori între mmol/l sunt suspecte, se repetă. Testul este necesar la toţi copiii cu tuse cronică. Pentru diagnosticul de FC: sensibilitate 98%, specificitate 83%, valoare predictivă pozitivă 99,5%. Testul genetic ARN: detectează care sunt genele defecte care au cauzat boala. Au fost identificate peste 1500 de mutaii genetice.

128
Testul sudorii fals-pozitiv 1. Mucoviscidoză 2. Malnutriţie severă 3. Etiologii endocrine Pseudo- hipoaldosteronismul Hipotiroidismul Hipoparatiroidismul Panhipopituitarismul Insuficienţă suprarenală 4. Etiologii metabolice Glicogenoză tip I Mucopolizaharidoză tip I 5. Altele –hipogamaglobulinemie –sindrom nefrotic –diabet insipid nefrogen –colestază familială –sindrom Klinefelter –anorexie mentală –nanism psihosocial –displazie ectodermică –maladia celiacă –deficit de G6PD –sindromul Mauriac Cîand un copil a fost diagnosticat cu fibroză chistică, atunci fraii i surorile trebuie să facă testul sudorii. Simptomele fibrozei chistice pot apare la vîrste diferite. Fraii i surorile pot avea fibroză chistică i aceasta să nu fi fost încă depistată.

129
Teste respiratorii EX RADIOLOGIC PULMONAR – pentru diagnosticul şi evoluţia manifestărilor pulmonare: îngroşări peribronşice difuze, atelectazii, emfizem pulmonar, bronşiectazii; TESTE FUNCTIONALE RESPIRATORII - Spirografia arată tulburări respiratorii tip obstructiv şi restrictiv; Scintigrafia pulmonară arată schimbări vasculare difuze (fibroză pulmonară). Modificări imunologice: hipogamaglobulinemia, nivele crescute de complexe imune. screening la nounăscui – între ziua a 5-a şi primele două luni de viaţă se face prin demonstrarea unui nivel crescut de tripsină imunoreactivă în sânge > 80 g/l (cel mai bun marker al depistării neonatale). Diagnostic prenatal: determinarea activităţii fosfatazei alcaline, enzime lizosomale în lichidul şi celulele amniotice din 18-20săpt.gest.; testul genetic ARN; ECHOgrafie prenatală din 11-12săpt. gestaie.

131
Diagnosticul diferenţial. Alte cauze de insuficienţă pancreatică exocrină: Sindromul Schwachman-Diamond. Boală rară, dar se consideră a doua cauză după mucoviscidoză. Boala este autosomal-recisivă, afectează pancreasul exocrin, măduva osoasă, creşterea metafizară. Insuficienţa pancreasului exocrin este datorată unei hipoplazii şi lipomatoze pancreatice. Se manifestă de la naştere prin diaree cronică, steatoree, malabsorbţie, malnutriţie severă. Concentraţia clorului î n sudoare este normală. Se asociază cu neutropenie cronică sau ciclică (cauză de infecţii severe recurente, frecvent cauză de deces precoce ), displazie metafizară şi nanism.

132
Pancreatita cronică -rară la copii. Steatoreea şi malabsorbţia apar la o extindere a leziunilor peste 80% din pancreasul acinar. Se confirmă prin ecografie abdominală, determinarea activităţii enzimatice î n sucul duodenal. Deficit izolat de lipază pancreatică. Boală autosomal-recisivă rară, deficitul de lipază determină malabsorbţia lipidelor. Scaunele au aspect de lichid uleios chiar de la naştere, dar fără repercusiuni severe asupra creşterii. Diagnosticul-prin dozarea lipazei î n sucul duoadenal. Celiachia, deficitul de dizaharidaze.

133
Tratamentul în fibroza chistică Tratamentul este complex, individualizat, continuă toată viaţa, în centre specializate. Obiectivele tratamentului FC: I.Terapia bolii pulmonare prin : Antibiotice pentru infecţia respiratorie Drenajul şi menţinerea permiabilităţii căilor respiratorii prin inhalarea de aerosoli, mucolitice, fiziokinetoterapie Tratamentul antiinflamator II.Terapia nutriţională: Regim igieno-dietetic Dieta bogată în proteine, calorii, redusă în lipide Substituţie cu enzime pancreatice Suplinirea cu vitamine, minerale III.Tratamentul complicaţiilor

134
Este foarte complicat... Pînă în prezent, nu există un tratament care să vindece boala, tratamentul existent fiind unul doar de ameliorare a simptomelor i de încetinire a evoluiei bolii!

135
TRATAMENTUL MANIFESTĂRILOR DIGESTIVE ABORDARE NUTRIIONALĂ Asistenţa nutriţională este necesară pentru restabilirea stării normale de nutriţie, reluarea creşterii ponderale, dar i pentru a influenţa suferinţa pulmonară din cadrul bolii, a îmbunătăţi calitatea vieţii şi prelungirea acesteia. Regimul igieno-dietetic prevede un aport energetic sporit cu 20-50% conform vârstei, circa kkal/kg corp în zi. Apariţia extractelor pancreatice sub formă de microsfere protejate de aciditatea gastrică permite o dietă hiperproteică, hiperglucidică, normală (sau chiar crescută?) pentru lipide. Dimensiunea mică a microsferelor permite tranzitul piloric, iar la nivelul duodenului proximal sub acţiunea pH-ului alcalin, învelişul enteric se dizolvă, eliberându- se enzimele pancreatice activate în circa 30 minute.

136
ABORDARE NUTRIIONALĂ Pentru sugari: formule lactate cu hidrolizate de proteine, trigliceride uşor asimilabile, polimeri de glucoză, vitamine şi minerale. Exemplu: Alfare (Nestle), Portagen(USA), Pregestemil, Humana (Germania); Clinutren -junior (Nestle) pentru copiii de 1-3 ani. Regim hidric de 2-3 litri pe zi. Pentru perioada caldă a anului –supliment de sare până la 1-4 grame în zi. Analiza regimului alimentar (împreună cu dieteticianul la fiecare 6 luni) Se recomandă consumul de glucide (bomboane, compoturi, îngheţată), brânzeturi, carne de pui, viţel, peşte, uleiuri vegetale Nu se va abuza de prăjeli, alimente pregătite pane, mezeluri, carne roşie grasă, ouă. Se contraindică consumul de viscere (creier, ficat etc), unt, bogate în colesterol. La nevoie (în caz de hipolipemie) se suplimentează cu preparate dietetice (Traumacal, Portagen, Fortimel etc)
, Portagen(USA), Pregestemil, Humana (Germania); Clinutren -junior (Nest")
137
ABORDARE NUTRIIONALĂ Supliment de vitamine liposolubile: Vit. A UI/zi la sugar UI/zi la copil Vit. D UI/zi Vit. E. 6-10mg/kg/zi Vit. K 3-5mg/i.m. lunar Vit. B 12 0,4 g/kg/zi Supliment de minerale: Fe, Zn, Se Fe: 2 – 3mg Fe elemental/zi Zn: 10 – 15mg Zn elemental/zi Se: 1 – 2 g/zi Supliment de NaCl: 1-2g/zi la sugar 2 – 4g/zi la copil

138
Terapia de substiuţie cu enzime pancreatice Este tratamentul estenţial al insuficienţei pancreatice Ameliorează pronosticul, calitatea vieţii, dieta cu un conţinut normal de lipide. Preparate: Creon, mezim-forte, pancitrat, pancreaza, prolipaza etc. Pentru copii mai frecvent utilizat Creonul UN cu 150mg pancreatină: amilaza-8.000, lipaza , proteaza- 600 UN (acum şi mezim de ). La adulţi Creon cu amilaza , lipaza , proteaza UN. Dozele medii sunt UN lipază la kg/corp la o priză alimentară sau UN lipază /kg corp/zi. Dozele mai mari cresc riscul stricturii de colon. Ulterior regimul dietetic este diversificat, ţinînd cont de toleranţa digestivă, tranzitul intestinal- toate pot fi influenţate de terapia de substituţie cu fermenţi.

140
Terapia de substiuţie cu enzime pancreatice La un copil sub 1 an: La 120 ml amestec 1/3-1/4 capsulă Kreon ( UN) Sau la 1 g lipide UN lipază, max UN lipază/kg/zi La copilul peste un an: 1-2 capsule la mesle de bază şi 1/2-1 capsulă la mese mici Doza nictimerală sub lipază/kg/zi sau UN lipază la 1g lipide pe zi Recomandări generale: Se utilizează doar fermenţii sub formă de microsfere. Fermenţii se utilizează la fiecare masă ce conţine lipide. Microsferele se amestecă cu puţine alimente (lapte matern) şi se administrează înaintea alimentaţiei (mai rar cu toată porţia)
 Sau la 1 g lipide 400-800 UN lipază, max 10.000 UN lipază/kg/zi La copilul peste un an: 1-2 capsule la mesle de bază şi 1/2-1 c")
141
Terapia de substiuţie cu enzime pancreatice De la 3-4 ani copilul înghite capsula la începutul sau la mijlocul mesei, este preferabilă administrarea 1/2 doză la începutul şi la mijlocul mesei. Doza se măreşte treptat pînă se obţine controlul manifestărilor clinice, caracterul scaunelor, adaosul ponderal, normalizarea absorbţiei (după coprogramă). Sugarul alimentat artificial se recomandă administrarea înainte şi după masă. Obiectivele: Un scaun, maximum două de aspect normal în fiecare zi; Absenţa durerilor abdominale; Coeficientul de absorbţie al grăsimilor 96 – 98%. Extractele pancreatice se administrează în cursul meselor, se evită zdrobirea capsulelor, la vârstă mică administrând-se doar conţinutul acestora, suspendat într-un lichid, hrană. Enzimele pancreatice trebuie luate la fiecare masă, tot timpul vieii!

142
TRATAMENTUL BOLII PULMONARE TRATAMENTUL INFECIEI CU ANTIBIOTICE. Este obiectivul terapeutic major. Are scop curativ în orice infecţie acută respiratorie, durata de circa 2-3 săptămâni, frecvent din start cu 2 antibiotice bactericide, administrare sistemică (i/m, i/v), doze medii- maximale. Tratamentul profilactic (de întreţinere) are scop de a suprima numărul şi activitatea germenilor, scăzând sursa antigenilor bacteriene care se află la originea complexelor imune (nu este unanim acceptat). Cure de tratament cu antibiotice intensiv aplicat de 3-4 ori /an (simestrial), timp de 2 săptămîni, de obicei i/v prin internare în centre specializate (nu în secţii de spital), dar acum majoritatea la domiciliu! (mai mult recomandabil în infecţia cu Pseudomonas aeruginosa). Antibioterapia este adaptată etiologiei cele mai frecvente: St.aureus, H.influenzae, P.aeruginosa, antibiogramei.

143
TRATAMENTUL BOLII PULMONARE Scheme eficiente recomandate: aminoglicozidele (gentamicina, tobramicina), penicilinele protejate (oxacilina, cloxacilina, amoxiclav, piperacilina, ticarcilina), cefalosporine de gen. III (ceftazidim, cefoperazon), fluorchinolone (ciprofloxacina, ofloxacina), vancomicina, În St. aureus: oxacilina, cloxacilina, amoxiclav, unasin,vancomicina, cefalexina, amonoglicozide în formele rebele sau la contraindicaţii pentru primele. În Haemophilus influenzae: amoxiclav, cefalosporine II-III, levomicetina, azitromicina. În Pseudomonas aeruginosa: cefalosporine (ceftazidim, cefoperazon), aminoglicozide, fluorchinolone. Se practică administrarea prin aerosol a antibioticelor, fie în cure separate, fie ca o completare la terapia enterală: colistină, gentamicină mg/zi, tobramicina mg/zi. Ceftazidim g/zi în priză unică (acţiune locală, prin nebulizare).
, penicilinele protejate (oxacilina, cloxacilina, amoxiclav, piperacilina, ticarcilina), cefalosporine de gen. III (ceftazidim, cefoperazon), fluorch")
144
Agenţi mucolitici. Este o componentă importantă a terapiei manifestărilor respiratorii. Preparatul de bază este N-acetilcisteina (thiole) sub formă de inhalaţii-nebulizări, oral, parenteral. Dozele 30 mg/kg/zi: 0-2 ani câte 50 mg de 3ori/zi, 2-6 ani câte 100 mg de 3-4 ori/zi, peste 6 ani câte 200 mg de 3-4 ori /zi Dozele prin inhalare sunt mai mici, de 2-3 ori /zi cu durata de 8-10 minute, cura până la 2 săptămâni. Alte mucolitice preferate: carbocisteina, mucosalvin, ambroxol, bromhexina. Bronhoscopie curativă cu lavaj traheo-bronşic rar, indicaţii stricte. În sindrom obstructiv – betamimetice (salbutamol) ca în astmul bronic.
 sub formă de inhalaţii-nebulizări, oral, parenteral. Dozele 30 mg/kg/zi: 0-2 ani câte 50 mg de 3ori/zi, 2-6 ani")
145
Fizioterapia-kinetoterapia. Esrte componentă importantă, indispensabilă a terapiei. Scopul principal-menţinerea permeabilităţii traheo-bronşice prin eliminarea adecvată a secreţiilor. Metode recomandate: Gimnastica respiratorie specială Drenajul postural (tapotare, vibraţie) Klopf-masajul Kineziterapia Inducerea tusei Inhalaţii şi nebulizări Efectele: stimularea tusei, mobilizarea-eliminarea secreţiilor, ameliorarea funcţiei respiratorii i funcţiei epiteliului ciliar, antrenarea musculaturii scheletale, ameliorarea calităţii vieţii, prevenirea infecţiei. Se petrece la domiciliu, la spital, în centre specializate sub supravegherea medicului, min.1-2 ore/zi Fiziokinetoterapia-agenţi mucolitici-terapia antibacteriană sunt utilizate în complex, îndepărtează progresarea bolii cronice pulmonare, determină calitatea, durata vieţii pacienţior cu FC.

146
Practicarea sportului. Activitatea fizică reprezintă o parte importantă în tratamentul FC. Practicarea sportului la persoanele cu FC previne deteriorarea plămînilor i intarete organismul, crete rezistena acestuia, crete masa musculară. Persoanele cu FC trebuie incurajate să practice divese forme de sport, în limita posibilităii fizice a acestora. Permise: înotul, alergări, bicicleta, tenis, badminton, schi, golf, voleibol, gimnastica, ioga, turismul. Nu se recomandă: patinaj, fotbal, hokei, bocs, regbi, basketbol, motosport, atletica grea, lupte diferite.

147
Tratamentul antiinflamator Pentru a stopa procesul inflamator imunopatologic bronşic şi fibroza. Sunt discutate antiinflamatoarele nesteroidiene (ibuprofen) şi steroidine (prednisolon oral şi inhalator). Tratamentul colestazei cu acid urso-deoxicolic.
 şi steroidine (prednisolon oral şi inhalator). Tratamentul colestazei cu acid urso-deoxicolic")
148
Tratamente contemporane i de viitor: Antiproteaze -alfa 1-antitripsina în aerosol, inhibă i reduce nivelul elastazei şi IL-K8 în mucusul bronşial (spută). Preparatul Pulmozim - DN-aze recombinante umane (rh DN-aze) în inhalaţii -neutralizează ADN-ul eliberat de celulele distruse în căile respiratorii, ameliorează funcţia respiratorie-mucolitic (este costisitor) Inhalaţii cu amilorid (diuretic) - blochează canalele de sodiu i scade reabsorbţia excesivă de sodiu, ameliorează vîscozitatea mucusului. Anticitochine, antiinterleuchine. Terapia genică, practicată din 1993: cu scopul de a întroduce în celulele submucoasei epiteliului respirator secvenţe de ADN cu CFTR normală () pe un vector viral (experiment pe adenovirus), poate corecta anomaliile de mişcare ale ionilor transmembranar. Transplantul de pulmoni sau cord-pulmoni rămîne de neînlocuit în faza terminală a bolii. Prognosticul este nefavorabil, durata de viată este de circa ani. În perspectivă pînă la ani. În RM durata de viaţă medie este de 10 ani, sunt unici pînă la 27 ani. Sunt importante psihoterapia, probleme medicosociale, crearea de centre specializate pentru bolnavi cu FC. Diagnosticul prenatal. Screeningul neonatal.
. Preparatul Pulmozim - DN-aze recombinante umane (rh DN-aze) în inhalaţii -neutralizează ADN-ul")
151
Dr. confereniar Petru Martalog Catedra Pediatrie nr.1 Deficitul de dizaharidaze

152
Intoleranţa la dizaharide Hidraţii de carbon (glucidele) din alimente sînt costituite din 2 grupe: glucidele şi fibre alimentare. Fibrele alimentare- sunt celuloza şi hemiceluloza, importante în procesul de evacuare a alimentelor şi în reglarea peristaltismului. Glucidele sînt substanţe ce se asimilează, asigură o producţie considerabilă din aportul caloric la copii (circa 50-60% ) constituind g/kg/zi. Glucidele alimentare se împart în monozaharide, dizaharide şi polizaharide. Dizaharide sunt constituite dinr-un rest de glucoză legat de un alt rest de fructoză (zaharoză), de galactoză (lactoză), de trehaloză, de celobioză.
 din alimente sînt costituite din 2 grupe: glucidele şi fibre alimentare. Fibrele alimentare- sunt celuloza şi hemiceluloza, importante în procesul de evacuare a alimentelor şi în reglarea peris")
153
ECFS Consensus Conference Artimino March 2004 Artimino, Italy March 2001

154
Glucide alimentare Proveni- enţa Compozi- ţia Digestia intralumi nală Digestia parietală Digestia în margine în perie Transport I.Polizaha ride 50-60% Vegetale Animale Amiloza Amilopecti na Dextrine glicogen α-amilaza salivară α-amilaza pancreas Enzime Glucoamil aza (maltaza) Dextrinaza izomaltaza Produşi finaliactiv II.Dizahari de 20% Vegetale Animale Zaharoza 30% Trehaloza Celobioza Lactoza 10% - Sucraza Trehalaza Lactaza Glucoza Fructoza Glucoza Fructoza Activ, maximum în jejun, minimum în ileon III.Monoza haride 10-20% Hexoze Pentoze Glucoza Frucoza Riboza Xiloza - - -Absorbţie directă, activă de la nivel stomacal

155
Enzimele, care hidrolozează dizaharidele sunt localizate în mucoasa în marginea în perie, activitatea maximală- jejun. Enzimele sunt aşezate stratificat astfel încît în imediata vecinătate a lumenului intestinal se află lactaza, apoi sucraza, izomaltaza, maltaza, trehalaza. Lactaza este cea mai importantă enzimă pentru sugar, deoacere este calea principală de asigurare a monozaharidelor, uor asimilabile la această vîrstă, localizarea ei superficială face ca activitatea să fie maximală. Hidroliza componentelor alimentare la acest nivel este strict specifică, de anumite enzime specifice. Monozaharidele finale sunt D-glucoză-80%; D-fructoză- 15%, D-galactoză- 5%. Procesul de absorbţie al monozaharidelor este activ, energodependent.

157
Dizaharidazele Îşi manifestă activitatea de la saptamîni intrauternin, nivel maxim la nou-născut la termen. Există lactaza copilului mic i lactaza adultului. Activitatea maximală este la sugarul mic, treptat scade pînă la 5 ani, după 5 ani se produce transferul cu sinteza lactazei de tip adult (este un proces genetic determinat) Putem avea o insuficienţă tranzitorie a lactazei la prematuri chiar şi la unii nou- născuţi la termen tip adult. Lactoza, este principalul dizaharid al laptelui. Lactoza este sintetizată în glanda mamară a tuturor mamiferelor (cu excepţia leului de mare). La toate mamiferele cantitatea de lactază este crescută conform nevoilor legate de alimentaţia exclusiv lactată. Odată cu înţărcarea nivelul lactazei scade semnificativ. Omul însă singurul mamifer care pastrează un nivel ridicat de activitate lactazică pînă la vîrsta de adult. Excepţie fac unele grupări etnice din zona africană i asiatică, Siberia- este o scădere considerabilă a activităţii lactazei dupa 4-5 ani i frecvenţa intoleranţei ajunge la 50-75%. La populaţia europeană frecvenţa este cca 10-20%. Acest comportament este programat genetic. Persistenţa activităţii lactazei la adulţi este privită ca o adaptaţie evolutivă la influenţele alimentare de mediu în procesul de selecţie naturală.

158
Lactaza, enzimă specifică, situată în marginea în perie intestinală realizează hidroliza lactozei în glucoză i galactoză. Lactaza este enzima principală în metabolismul DZ la copilul sugar; dar ea este cel mai superficial localizată i este cel mai frecvent afectată. Absenţa sau reducerea activităii dizaharidazelor determină perturbarea digestiei i absorbţiei hidrocarboonatelor respective. Dizaharidele nemetabolizate în intestinul subţire nu se asimilează, pot declanşa un tablou clinic clasic – tipic, definit ca intoloeranţă la dizaharide în care simptomul principal este diareea cronică specifică. Intensitatea simptomelor depinde de cantitatea de dizaharide ingerate, de motilitatea gastro-intestinală, activitatea florei bacteriene intestinale. Odată declanşată, diareea i malabsorbţia glucidelor determină un dezichilibru hidroelectrolitic şi caloric, care intr-o primă etapă pot duce la deshidratarea acută cu punerea in primejdie imediată a vieţei sugarului. Persistenţa fenomenelor în timp au ca consecinţă diareea cronică, malabsorbţia, oprirea creşterii staturo-ponderale.

159
Clasificarea. Malabsorbţia polizaharidelor: I. Malabsorbţia congenitală (primară) a amidonului. II. Malabsorbţia dobândită (secundară) a amidonului. Malabsorbţia dizaharidelor: I. Deficitul de lactază: Deficitul congenital (primar) de lactază: cu lactozurie (tip Holzel), fără lactozurie (tip Durând), tip tardiv. Deficitul dobândit (secundar) de lactază. II. Deficitul de sucrază-izomaltază. III Deficitul congenital de trehalază. Malabsorbţia monozaharidelor: I. Deficitul congenital (primar) de glucoză-galactoză. II. Deficitul fructozei. III Deficitul secundar de glucoză-galactoză-fructoză.
 a amidonului. II. Malabsorbţia dobândită (secundară) a amidonului. Malabsorbţia dizaharidelor: I. Deficitul de lactază: Deficitul congenital (primar) de lactază: cu lac")
160
Deficite secundare a lactazei - cauze: leziuni ale mucoasei intestinale în infecţi intestinale virale, bacteriene; unii autori confirmă hipolactazia în 100% infecţie rotavirală; 30-45% după dizenterie, salmoneloză, colienterite. alergia alimentară a copilului mic atrofia mucoasei intestinale în celiachie, alte malabsorbţii helmintiaze- giardiaza (lamblioza) ş.a. medicamente: antibiotice (neomicină, canamicină, ampicilină), tratament prelungit cu citostatice; unele anabolice, substanţe narcotice intoxicaţii acute şi cronice prematuritatea rezecţii intestinale tranzit intestinal accelerat sindromul poluării bacteriene excesive intestin Are loc reducerea numărului de enterocite, scăderea capacităţii acestora de a sinteza lactoza (reducerea suprafeţii de absorbţie). Important. Toate aceste maladii duc la malnutriţie protein – calorică. Aceasta, la răndul său, la fel poate determina o hipolactazie secundară (cerc vicios).

161
Fiziopatologie În normă monozaharidele rezultate din hidroliza dizaharidelor sînt trasportate activ în celule. Laptele uman conţine lactoză în cantitate de 7% şi cel bovin 4,8%, asigurînd circa 55% şi 40% din raţia calorică zilnică a sugarului. Lactoza este hidrolizată de lactază în glucoză şi galactoză. Lactoza (toate dizaharidele) nehidrolizată practic nu se absoarbe în intestine şi determină o alterare succesivă, profundă a metabolismului intestenal.

162
Dizaharidele nedigerate cresc concentraţia osmotic–activă în intestine, cu o atragere de apă i electrolii din spaţiile interstiţiale spre lumenul intestinal cu diaree apoasă Dizaharide nehidrolizate ajung în colon, sînt parţial sau complet degradate sub acţiunea dizaharidazelor bacteriilor intestinale. Ca urmare se produce o acumulare de acizi organici (acid lactic, acid acetic), atomi de hidrogen, bioxid de carbon, apă. Toate au efecte iritative locale asupra mucoasei (excitomotor şi excitosecretor), ce generează hiperperistaltism cu accelerarea tranzitului intestinal. Acumularea de CO2 – duce la meteorism intestinal marcat, dureri abdominale (colici). Acidul lactic si acetic cresc osmolaritatea, scade pH-ul intestinal, ce la rîndul lor perturbă şi mai mult absorbţia lichidelor şi electroliţilor în colon, la fel irită mucoasa intestinală (pH-ul scaune sub 6,0). Surplus de dizaharide, de fibre vegetale – creşte volumul maselor fecale, ce şi mai mult cresc tranzitul intestinal (cerc vicios!)

163
Consecinţe: Diaree cu scaune frecvente (8-10 ori/zi), spumos-apoase, miros acriu (pH sub 5.5), voluminoase, emisie explozivă (sub presiune); culoare galben-deschisă. pH-ul acid irită pielea fesieră cu eritem. Copilul are vărsături, refuză alimentaţia, prezintă meteorişm cu borborisme intestinale. Diareea persistă o perioadă oarecare chiar şi după întreruperea alimentaţiei lactozate. La copil mic - pierdere considerabilă de apă, electroliţi, cu sindom de deshidratare acută. Secundar acestor modificări se produce: Creşterea excreţiei de proteine (exudaţie) la nivel intestinal prin alterarea epiteliului intestinal, prin modificarea calibrului vascular- se pierd proteine plasmatice cu hipoproteinemie neselectivă; Hipocalcemie prin pierderea efectului favorabil al lactozei în absorbţia de Ca, necesitatea excluderii laptelui; Hiposideremie; Lactozurie – alterarea peretelui intestinal face posibilă trecerea în circulaţia sanguină a unei cantitaţi de lactoză prin difuzie pasivă. Rar disfuncţii renale tubulare şi stenoză pilorică.
, spumos-apoase, miros acriu (pH sub 5.5), voluminoase, emisie explozivă (sub presiune); culoare galben-deschisă. pH-ul acid irită pielea fesieră cu eritem. Copilul are vărsături, refuză alimentaţia")
164
Intoleranţa la lactoză Deficitul lactazei este o maladie primară (ereditară) sau secundată (dobândită) Cauza - incapacitatea totală (alactazie) sau parţială (hipolactazie) a enterocitelor de a sintetiza lactaza. Este desemnată ca cea mai frecventă cauză de malabsosrbţie la copil Deficitul de lactază se manifestă în 15-30% din cazuri la copiii cu o simptomatică abdominală şi la 45,7% dintre copii aparent sănătoşi, cu vârste cuprinse între 5 şi 12 ani.
 sau secundată (dobândită) Cauza - incapacitatea totală (alactazie) sau parţială (hipolactazie) a enterocitelor de a sintetiza lactaza. Este desemnată ca cea mai frecventă ca")
165
INTOLERANŢĂ CONGENITALĂ LA LACTOZĂ (ALACTAZIA SUGARULUI) Deficit ereditar de lactază, tip Holzel (1959), transmitere autosomal-recesivă. Incidenţa este foarte rară. Debutul poate fi chiar din primele mese cu lapte ale copilului. Vărsăturile sunt constante, apoi rapid urmate de diaree caracteristică: scaune frecvente, apoase, spumoase, acide, explozive, însoţite de distensie abdominală, meteorism, stare de agitaţie. Ca consecinţă, apar pierderea în greutate, deshidratare pronunţată, toxicoză, instalarea unei malabsorbţii. Dacă nu se sistează alimentaţia cu lapte, este posibil decesul. Invers, anularea alimentaţiei cu lapte rapid duce la ameliorarea stării, creşterea în greutate, etc. Diagnosticul pozitiv: istoricul bolii, scaune acide cu pH scăzut, testul cu H2 expirat pozitiv, testul de toleranţă la lactoză pozitiv, surplus de lactoză- curba glicemică plată. Lactozuria este caracteristică. Testul de bază ar fi biopsia mucoasei intestinale: arată o arhitectură normală cu absenţa activităţii fermentului lactaza. Altă formă – alactazia tip Durand – este o formă mai gravă. În afara lipsei lactazei, este crescută permeabilitatea mucoasei intestinale cu creşterea lactozei serice, efecte toxice ale lactozei asupra SNC şi rinichilor. Mai frecvent se întîlneşte la familii înrudite. Debut precoce, evoluţie gravă cu deces frecvent. Sînge – lactozemie. Aminoacidurie. Caracter familial, autosomal-recesiv.
 Deficit ereditar de lactază, tip Holzel (1959), transmitere autosomal-recesivă. Incidenţa este foarte rară. Debutul poate fi chiar din primele mese cu lapte ale copilului. Vărsăturile sunt cons")
166
Deficitul secundar de lactază (E73.1) Tabloul clinic este în funcţie de maladia de bază, vîrsta, gradul de lezare a mucoasei intestinale, gradul de suprimare a secreţiei de lactază, cantitatea produselor lactate din alimentaţie. Debutul este treptat, semnele de bază sunt diareea apoasă, deshidratarea, malnutriţia. Diareea din intoleranţa la dizaharide are caracter particular: diareea apare în primele 30 min. după alimentaţie. Scaune frecvente, apoase, spumoase, se emit exploziv, culoare galben deschisă, miros acru (pH acid), apare eritem fesier (aciditate). Diareea este însoţită de vome repetate, refuzul alimentaţiei, meteorism, borborisme, flatulenţă, dureri abdominale (frecvent aceste semne precedă diareea). Diareea este abundentă şi rebelă, poate duce la o deshidratare şi toxicoză. Diareea este persistentă, chiar şi o perioadă de timp după întreruperea lactozei. Deficitul neonatal de lactază tranzitoriu apare precoce, frecvent la prematuri, caracteristic diareea apoasă, apare la min. după alimentaţie, este precedată frecvent de meteorism, borborisme, copilul lasă sînul, este neliniştit, agitat, emite scaune apoase, explozive, acide, spumoase, după care se linişteşte, reia alimentaţia. Statutul ponderal corespunde vîrstei, se corectează cu vîrsta (de obicei către 2-4 săpt.).
 Tabloul clinic este în funcţie de maladia de bază, vîrsta, gradul de lezare a mucoasei intestinale, gradul de suprimare a secreţiei de lactază, cantitatea produselor lactate din alimentaţie. Debutul este treptat,")
167
Deficitul de lactază tip tardiv este formă cu micşorarea activităţii lactazei după limitarea aportului de produse lactate şi/sau după substituirea lor prin alte echivalente nutritive. Varianta a fost descrisă în Din totalul formelor deficitului de lactază acestui tip îi revine o rată de 11%. enzima responsabilă se sintetizează fie în cantităţi scăzute, fie are o activitate redusă. Defectul genetic presupus micşorează sinteza precursorului în reticulul endoplasmatic şi alterează procesul posttranslaţional. In rezultat enzima, acumulându-se în aparatul Golgi, ulterior este degradată la nivelul lizozismelor. Cu toate că este posibilă şi o degradare prematură a enzimei. Odată cu întreruperea alăptării, datorită unor implicări fiziologic- adaptative, lactază îşi diminuează activitatea sa la 5-10% din nivelul prezent la naştere. Numeroase studii etnografice au stăruit asupra faăptului că această stare, în cadrul anumitor grupuri etnice, indiferent de arealul habitat, apare cu o frecvenţă de până la 90%, fiind definită ca defect genetic-adaptativ în gena responsabilă de reglarea sintezei lactazice, transmisă pe cale autosomal dominantă.

168
Diagnosticul Examenul coprologic: pH acid sub 6.0, osmolaritatea crescută peste 40 mEq/l, acizi graşi, floră iodofilă. Testul de toleranţă la glucoză nu este schimbat. Testul de toleranţă la lactoză – administrare orală a 1-2 g/kg de lactoză şi apă cu aprecierea glicemiei şi galactozemiei. Testul este pozitiv cînd la scurt timp apare diareea apoasă, galactozemia apreciată la fiecare 30 min. nu depăşeşte valorile iniţiale cu 0, 5 mg/l, iar glicemia cu 2 mg/l – curba plată. Aprecierea H2 în aerul expirat – o creştere după dejun de probă cu lactoză. Analiza histoenzimatică a bioptatului jejunal – nivelul lactozei redus sub 15 UI/g Teste de excludere

169
Diagnosticul diferenţial I ntoleranţa la proteinele laptelui de vaci – apare numai la introducerea laptelui de vaci, se asociază cu fenomene alergice, vărsături frecvente, scaune diferite, copilul tolerează laptele matern. Deficit de alte dizaharidaze.

170
TRATAMENTUL Este dietetic, cu excluderea sau reducerea din alimentaţie a preparatelor de lapte ce conţin lactoză. În formele congenitale confirmate, lactoza se exclude pe toată viaţa. În formele secundare se sistează preparatele din lapte pentru luni (lactază îşi restabileşte activitatea încet). Alimentaţia sugarului se face cu preparate din lapte delactozate, sau hipolactozate. Tratamentul vizează suplinirea deficitului glucidic prin alte hidrocarburi, echilibrarea deficitului calcic, administrarea substituienţilor enzimatici. Trebuie identificate şi excluse toate produsele ce conţin lapte dismulat sau lapte praf ( bomboane, ciocolată, conserve, salamuri). Alimentaţia se va face cu preparate pe bază de soia, monozaharide şi glucoză.

171
În perioada de excludere raţia alimentară a sugarilor se constituie din substituenţi de lapte pe baza hidrolizatelor proteice. Considerând că majoritatea micuţilor nu acceptă aceste produse din cauza calităţilor gustative, sunt recomandate amestecurile delactozate şi substituenţii din soia. Reintroducerea produselor lactate se face fracţionat, începând cu produse acidolactice. Raţia alimentară a acestor copii se lărgeşte cu legume şi cu fructe (sub forma omogenizatelor comercializate în borcănaşe mici), carne, puţin unt. După respectarea acestui regim, perioadă de un an, se permit produse de patiserie care nu conţin lapte. Este importantă evitarea consumului accidental de lactoză. De aceea, procurând produse pentru copiii cu un astfel de regim, eticheta de însoţire! trebuie studiată cu prudenţă. La prescrierea medicamentelor se ţine cont ca acestea să nu conţină în excipienţii săi lactoză. Pentru copiii mai mari, diversificarea regimului fără lactoză este apropiat regimului fără proteinele laptelui de vacă, spre deosebire de care se permite carnea de vită.

172
În deficitul secundar, deficitul tranzitoriu de lactază sunt folosite preparatele hipolactozate, administrate doar pe o perioadă de timp pînă la refacerea enterocitelor. În aceste forme sunt permise brînzeturile fermentate şi proaspete, iaurturile (sunt tolerate datorită bacteriilor fermentative), produse delactozate NESTLE: Alfare, Nan fără lactoză, Clinutren junior. Pentru sugarul mic alimentat natural se poate încerca ½ -1/3 din volum de amestec delactozat, apoi lapte matern. După 5 luni – produse din soia, după 6 luni – brînză, unt, caşcaval. Peste hotare sunt preparate care scindează lactoza din lapte şi atunci laptele este tolerat bine. Preparatele Kerulac, Lactozoenzim, picături sau capsule se amestecă cu laptele, se incubează 2-3h şi se foloseşte. Reintroducerea produselor lactate se face fracţionat sub supravegherea medicului. Episoadele de deshidratare se tratează în funcţie de severitatea pierderilor. Este importantă suplinirea ratei cu calciu, tratamentul anemiei, rahitismului, malnutriţiei. Pentru diversificare sunt produsele Nestle. De la 6 luni terci de orez, hrişcă, ovăz, 3 cereale, 8 cereale. Pireuri din fructe (prune, măr, pere, bostan); De la 8-9 luni legume cu carne de pui sau de vită, Copilul de la 12 luni – Clinutren junior Evoluţia bolii este favorabilă.

173
Исключаются РазрешаютсяНазвание СтранаФирмаHa 100 мл готовой смеси белки, г жиры, г угле воды, г ккал Женское молоко. Все молочные продукты и смеси Безлактозные молочные смеси НАН безлактоз рый ШвейцарияNestle1,93,37,467 Bebelac-FLГолландияLijempf1,73,468,769 Portagen HiPP Nutrilon США MeadJo hnson 2,43,27,966,7 Адаптированны е соевые смеси Enfamil- Soy США Mead Johnson 2,03,66,567 Humana- SL ГерманияHumana2,03,67,972 Nutri-Soja ГолландияNutricia1,83,66,767 Frisosoy Голландия 1,73,57,167 Alsoy ШвейцарияNestle1,93,37,467 Similac- Isomil США Ross Labo ratories 1,83,76,868 Soja-Semp ШвецияSemper2,03,46,966 Heinz-Soy Formula СШАHeinz1,953,87,070 Продукты, исключаемые и рекомендуемые при алактазии

175
Dr. confereniar Petru Martalog Catedra Pediatrie nr.1 Intoleran ţ a la proteinele laptelui de vac ă

177
Intoleranţa la proteinele laptelui de vacă (IPLV) – este o hipersensibilitate de tip imuno-alergic la proteinele laptelui de vacă cu manifestări digestive, cutanate, respiratorii, care dispar la sistarea laptelui de vacă din alimentaţie. IPLV este considerată o cauză importanţa de diaree cronică a sugarului sub 6 luni. Incidenţa generală este 0,5-7 % la copii sub 2 ani.
 – este o hipersensibilitate de tip imuno-alergic la proteinele laptelui de vacă cu manifestări digestive, cutanate, respiratorii, care dispar la sistarea laptelui de vacă din alimentaţie. IPLV este co")
178
Etiologie. Patogenie. Factorul etiologic determinant este proteina din laptele de vacă (3,4g/100 ml) Fracţiunile cu capacitate alergizantă mare sunt beta- lactoglobuline, cazeina, alfa-lactalbumina, serum-globulina. În mod normal, mucoasa intestinală nu este permeabilă pentru structuri proteice nedigerate. Proteinele pot fi absorbite numai sub formă de aminoacizi, oligopeptide. În condiţiile unor imperfecţiuni structurale (nou-născut, sugar), are loc creşterea permeabilităţii intestinale pentru macroproteine. Betalactoglobulina este rezistentă la temperaturi înalte, la pH acid, este greu digerabilă, absorbţia intestinală se face sub formă de molecule intacte. Are loc sensibilizarea mucoasei intestinale cu răspuns ulterior imuno-alergic sistemic specific.
 Fracţiunile cu capacitate alergizantă mare sunt beta- lactoglobuline, cazeina, alfa-lactalbumina, serum-globulina. În mod normal, mucoasa intestinală")
179
Etiologie. Patogenie. IPLV pote evolua după 3 tipuri de reacii imune Tipul I (imediat, anafilactic, reaginic) implică anticorpi tip ig E fixaţi pe mastocite sensibilizate, este forma cea mai frecventă, apare imediat după ingestie. Tipul III (prin complexe imune). Prin formare de comlexe imune din antigene alimentare şi anticorpi IgM, IgG, reacţia se petrece la nivelul mucoasei intestinale, activarea complementului, eliminare de mediatori ai inflamaţiei. Semnele clinice apar la 6-8 ore după ingestie LV, dureză săptămîni. Tipul IV (mediat celular, tip întîrziat). Leziunile sunt mediate de limfocite T citotoxice, infiltrat limfocitar al mucoasei ce induc atrofie intestinală. Apar tardiv la 2-10 zile după ingestie, evoluţie cronică.
 implică anticorpi tip ig E fixaţi pe mastocite sensibilizate, este forma cea mai frecventă, apare imediat după ingestie. Tipul III (prin com")
180
Factorii predispozanţi imaturitatea imunologică (lipsa IgA) şi histologică a mucoasei intestinale la nou-născuţ şi sugar mic; teren individual atopic cu nivele crescute de ig E; istoric familial de alergie-atopie, inclusiv IPLV; Agresiune infecţioasă intestinală; Se discută o modalitate de transmitere autosomal- dominantă a IPLV. Anatomie patologică Afectare duodeno-jejunală. La nivelul mucoasei intestinale: infiltrat cu limfocite, plasmocite, eozinofile, glandele hiperplazate. Histologic-atrofie parţială sau totală vilozitară, uneori aplatizare vilozitară.
 şi histologică a mucoasei intestinale la nou-născuţ şi sugar mic; teren individual atopic cu nivele crescute de ig E; istoric familial de alergie-atopie, inclusiv IPLV; Agresiune infecţioasă")
181
Manifestările clinice Debutul maladiei este în primele 6 luni de viaţă, cu vîrste limitate între prima zi de viaţă şi după 12 luni. Semne de apel, de alarmă: apariţia tulburărilor digestive cu caracter postprandial la sugarul mic după întroducerea LV, rar peste 1-2 luni; apariţia semnelor clinice în deplină stare de sănătate; constatarea unei componente anafilactoide (paloare, tahicardie); reapariţia diareei la reluarea alimentaţiei cu LV.

182
Manifestările clinice Formele acute apar imediat după alimentaţie. Clinic evoluţie tip gastroenterită. Diareea apare în % cazuri, este precedată de colici abdominale, caracter postprandial. Scaune frecvente, apoase, lichide, explozive, meteorism şi deshidratare. Vărsăturile sunt prezente la circa 70-80% cazuri, pot fi unice sau repetate, frecvent preced diareea şi corelarea lor cu ingestia LV este uşor de stabilit. De obicei vărsătura se asociază cu un scaun lichid omis în get, paloare care dispar la cîteva ore şi reapar la următoarea alimentaţie. Forma de tip anafilactic. Apare după un biberon cu LV, adesea parţial refuzat, brusc cu paloare, 1-2 vărsături repetate, cianoza periferică, tahicardie, hipotonie, tulburări de conştiinţă, laringospasm, şoc.

183
Manifestările clinice Forma acută hemoragică - formă rară cu vărsături cu striuri de sînge pînă la hematemeza veritabilă, scaune sanguinolente, anemie. Forme cronice ale IPLV. Au debut insidios, după întroducerea în alimentaţie a LV. Semnul clinic este diareea cronică, stabilirea sindromului de malabsorbţie cu steatoree şi creatoree, malnutriţie. Periodic crize de deshidratare. La examenul clinic abdomen metiorizat, mare în contrast cu extremităţile subţiri (ca în celiachie). Caracteristic anorexie, dureri abdominale, diaree rebelă gravă, extravazări de sînge şi plasmă (sînge ocult în fecale), uneori puroi. Forma cu tablou clinic de enteropatie exudativă. Apare în evoliţia cronică a IPLV. Caracreristic diaree rebelă, extravazare de sînge şi plasmă în scaune, hipoproteinemie, eozinofilie, edeme, ascită.

184
Manifestări extradigestive în IPLV Frecvent sunt asociate manifestărilor digestive, rar sinestătător. Fenomene de şoc anafilactic: paloare, confuzie, hipotonie, cianoză periferică, şoc. Fenomene respiratorii: rinita alergică, bronşita obstructivă, wheezing, infiltrate pulmonare, otită. Fenomenele cutanate: edem al feţei pînă la edem Quinche, prurit, dermatită, exemă.

185
Diagnosticul IPLV Anamneza. Apariţia primară a semnelor clinice la întroducerea precoce a LV; Dispariţia semnelor clinice la excluderea LV i reapariţia lor la ore-zile după reîntroducerea LV. Examenul coprologic: prezenţa de eozinofile, cristale Charcot-Leyden; pot fi leucocite şi puroi; microhemoragii oculte- proba Gregersen pozitivă. În sindromul de malabsorbţie - alimente nedigerate, steatoree, amiloree, creatoree moderate. Anemie hipocromă, feriprivă, eozinofilie. Hipoproteinemie, hipoalbuminemie, hipocalciemie.

186
Diagnosticul IPLV Teste imunologice (nu sunt specifice, importanţă minoră): Nivele brusc crescute de anticorpi anti-PLV (precipitine, hemaglutinine) cu titru peste 1:512 la circa 75% cazuri (Ig M, Ig G) Nivele crescute de ig E - la circa 25% Testul de transformare limfoblastică - cel mai valoros, pozitiv la 75% bolnavi, testul pozitiv la betalactoglobulină, cazeină, alfalactalbumină Testele cutanate la antigenele alimentare pozitive. Biopsia intestinală: atrofie vilozitară parţială, lamina propria edemată, infiltrată cu eozinofile, plazmocite, limfocite (test neconcludent şi nenecesar!) Testul de provocare.
: Nivele brusc crescute de anticorpi anti-PLV (precipitine, hemaglutinine) cu titru peste 1:512 la circa 75% cazuri (Ig M, Ig G) Nivele crescute de ig E - la circa 25% Testul d")
187
Diagnosticul diferenţial. Necesită excluderea bolilor cu debut precoce cu diaree cronică, malabsorbţie, malnutriţie: intoleranţa la dizaharide, celiachie. Enterocolitele acute, alergia alimentară.

188
Tratamentul. Tratamentul de bază al IPLV este excluderea laptelui de vaci din alimentaţia sugarului. La copilul mic - laptele matern. Produsele ideale -amestecurile adaptate în baza hidrolizatelor proteice: Progestemil, Alfare, Nutramigen, Nutrilon care nu prezintă risc de sensibilizare. Partea minus-cost mare, osmolaritate crescută, gust prost. Alternativa - amestecuri în baza proteinelor din Soia: Nutrisoia, Nutrilon-Soia, NAN-Soia, Humana etc. Este dovedită prezenţa alergiei încrucişate şi la proteinele din soie la circa % copii cu IPLV.

189
Tratamentul. La copil după 4-6 luni: crupe pe apă, legume-fructe, ulei vegetal, produse din carne (exclus carne de vită). În formele grave, cronice - la etapa iniţială se exclude glutenul, lactoza. La prezenţa complicaţiilor severe - alimentaţia parenterală. La copii cu IPLV nu se recomandă fermenţi pancreatici din pancreas de bovine-pot accentua diareea. Suplinirea de vitamine, microelemente (vit.D, Ca, Fe). Evolutia. În formele acute excluderea LV are efect aproape imediat -cu dispariţia semnelor clinice. Remisiunea clinică apare în 1-2 săpt, reluarea creşterii ponderale în 2-4 săpt., normalizarea creşterii - în cîteva luni. Vindecarea histologică este lentă, revine la normal în 6-12 luni.
. În formele grave, cronice - la etapa iniţială se exclude glutenul, lactoza. La prezenţa complicaţiilor severe - alimentaţia pare")
190
Tratamentul medicamentos. În stare de urgenţă - tratamentul şocului anafilactic; Terenul atopic-antihistaminice gen.II (claritin, loratidina, cetirizină), tratament local; Inhibitori ai degranulării mastocitelor: cetotifen, nalcrom, cetirizina pentru 6-8 săpt.; Forme cronice: suplinire de minerale, vitamine, tratamentul malnutriţiei, deshidratării. Reîntroducerea laptelui de vacă. Se recomandă după vîrsta luni, rar toleranţa se obţine după 4-6 ani. Reîntroducerea LV se face treptat, de la 5- 30ml/zi, de dorit în condiţii de spital, riscul şocului neputînd fi niciodată îndepărtat. Pronosticul este bun.
, tratament local; Inhibitori ai degranulării mastocitelor: cetotifen, nalcrom, cetirizina pentru")








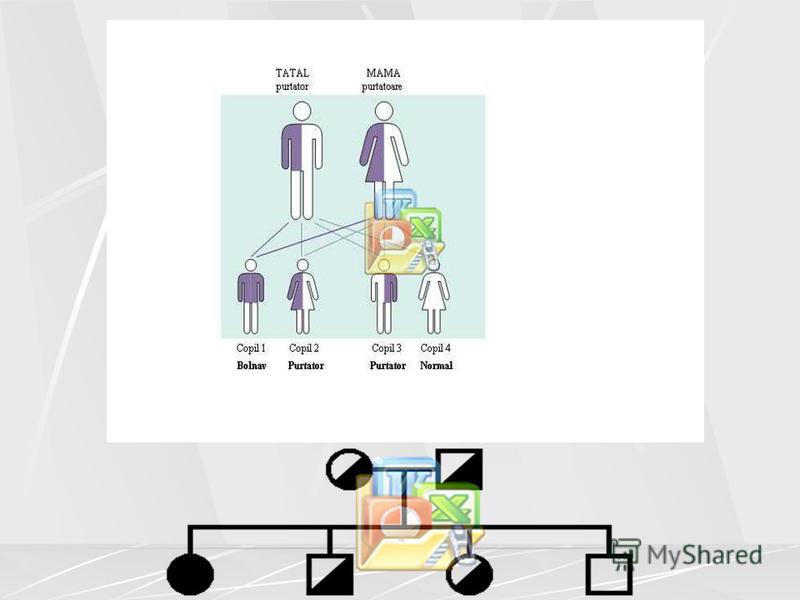









 După tipul de hemoliză Streptococii beta-hemolitici (produce.")
")


 Starea solidă: au formă proprie; rigiditate; au volum propriu (incompresibilitate). b) Starea lichidă:")














