Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
Metabolismul glucidelor

2
În alimentaţie glucidele sunt reprezentate de: polizaharide (60%), dizaharide (30)% şi monozaharide. În alimentaţie glucidele sunt reprezentate de: polizaharide (60%), dizaharide (30)% şi monozaharide. Monozaharidele pot fi absorbite ca atare, di – şi polizaharidele se supun în prealabil hidrolizei. Monozaharidele pot fi absorbite ca atare, di – şi polizaharidele se supun în prealabil hidrolizei. În cavitatea bucala amilaza salivară iniţiază digestia glucidelor, care va continua în stomac, aici enzima este protejată un anumit timp de amidonul îngerat. În cavitatea bucala amilaza salivară iniţiază digestia glucidelor, care va continua în stomac, aici enzima este protejată un anumit timp de amidonul îngerat.
, dizaharide (30)% şi monozaharide. În alimentaţie glucidele sunt reprezentate de: polizaharide (60%), dizaharide (30)% şi monozaharide. Monozaharidele pot fi absorbite ca atare, di – ş")
3
În stomac polizaharidele se transformă în proporţie 35 – 48 %. În stomac polizaharidele se transformă în proporţie 35 – 48 %. Amilaza salivară la nou-născuţi are rol important la insuficienţa secreţiei de amilază pancreatică, de alt fel ca şi la bolnavii de pancreatită cronică. Amilaza salivară la nou-născuţi are rol important la insuficienţa secreţiei de amilază pancreatică, de alt fel ca şi la bolnavii de pancreatită cronică. În intestin continuă hidroliza glucidelor sub acţiunea amilazei pancreatice până la: maltoză, maltotrioză şi α – dextrine. În intestin continuă hidroliza glucidelor sub acţiunea amilazei pancreatice până la: maltoză, maltotrioză şi α – dextrine.

4
Digestia are loc în marginea în perie a membranei enterocitale, enzimele nu sunt secretate în sucul intestinal. Maltoza, maltotrioza, α – dextrinele, dizaharidele îngerate străbat prin difuzie membranele enterocitelor, unde se va desăvârşi digestia. Există 7 dizaharidaze care desăvârşesc digestia - complexele: sucraza-izomaltaza, maltaza- glicoamilaza, lactaza-florizin hidrolaza şi trehalaza.

5
α – dextrinele sunt scindate de 1,6 – glucozidaza (dextrinaza). Trehalaza hidrolizează trehaloza – dizaharid rar întâlnit în alimentaţie. Glucozidazele prezintă adaptare în funcţie de regimul alimentar – lactaza este activă la nou- născuţi şi dispare după întreruperea regimului de alimentare.
. Trehalaza hidrolizează trehaloza – dizaharid rar întâlnit în alimentaţie. Glucozidazele prezintă adaptare în funcţie de regimul alimentar – lactaza este activă la nou- născuţi şi dispare")
6
Absorbţia glucidelor: Monozaharidele rezultante (glucoza, galactoza, fructoza) sunt eliberate în vecinătatea imediată a sistemelor de transport. Sunt necesari ionii Na +, Na + /K + ATP-aza – care eliberează energia necesară pentru absorbţie. Procesul de absorbţie este activ prin fosforilare.
 sunt eliberate în vecinătatea imediată a sistemelor de transport. Sunt necesari ionii Na +, Na + /K + ATP-aza – care eliberează energia necesară pentru absorbţie. Procesul")
7
Fosforilarea este stimulată de vitaminele B 1, C, metionina şi hormonii suprarenali. Transportorul leagă în locuri separate glucoza şi Na +, glucoza părăseşte celula prin difuzie facilitată, Na + este expulzat contra gradientului de concentraţie prin intervenţia enzimei Na + /K + ATP-aza. Anabaena inhibă enzima şi respectiv transportul glucozei.

9
Monozaharidele ajung prin vena portă la ficat. O parte se transformă prin gliconeogeneză în glicogen, iar alta (doar glucoza) trece în circulaţie. Galactoza şi fructoza sunt transformate în glucoză.
 trece în circulaţie. Galactoza şi fructoza sunt transformate în glucoză.")
10
Glucoza sanguină este utilizată pentru: necesităţi energetice; necesităţi energetice; transformare la nivelul muşchilor în glicogen. transformare la nivelul muşchilor în glicogen.

11
Absorbţia glucidelor depinde de prezenţa: Amilazei şi a unei funcţii pancreatice normale. Dizaharidazelor la nivelul enterocitului. Mucoasei intestinale normale cu mecanisme de transport active normale.

12
Transferul intracelular al glucozei: Străbate membrana celulară în ambele sensuri fără consum de energie cu ajutorul unor transportatori pasivi. Transportatorii glucozei (Glu T) sunt o familie de glicoproteine transmembranare, codificate de diferite gene.
 sunt o familie de glicoproteine transmembranare, codificate de diferite")
13
Fixarea glucozei pe faţa extracelulară a membranei determină o modificare conformaţională a proteinei transportatoare, facilitând trecerea şi eliberarea glucozei în interiorul celulei. Exprimarea genetică a Glu T este dependentă de tipul de ţesut şi se deosebesc între ei prin afinitatea pentru glucoză, exprimată prin Km.

14
Tipurile de transportatori Glu T: a) G luT 1 sunt preponderenţi la nivelul eritrocitelor, placentei; Km 1 mmol, concentraţie inferioară celei de glicemie, favorizând intrarea glucozei în celule, chiar în condiţii de hipoglicemie, în perioadele dintre mese. b) G luT 2 sunt preponderenţi în ficat şi pancreas, Km se cuprinde între 15 – 20 mmol, concentraţie superioară celei de glicemie postprandială, ceea ce determină intrarea rapidă a glucozei provenite din absorbţia intestinală în hepatocite, în condiţii de hiperglicemie; în caz contrar, în situaţii de hipoglicemie pătrunderea glucozei în hepatocite este minimă.
 G luT 1 sunt preponderenţi la nivelul eritrocitelor, placentei; Km 1 mmol, concentraţie inferioară celei de glicemie, favorizând intrarea glucozei în celule, chiar în condiţii de hipoglicemie, în perioadele dintre")
15
c) GluT 3 sunt preponderenţi la nivelul encefalului, placentei şi au acelaşi caracteristici ca şi GluT 1; d) GluT 4 sunt preponderenţi la nivelul ţesutului adipos şi muscular; Km 5 mmol, valoare apropiată de cea a glicemiei; sinteza şi afinitatea lor pentru glucoză este reglată de către insulină; e) GluT 5 sunt prezenţi în special la nivelul epiteliului intestinal, unde intervin şi în transportul fructozei.
 GluT 3 sunt preponderenţi la nivelul encefalului, placentei şi au acelaşi caracteristici ca şi GluT 1; d) GluT 4 sunt preponderenţi la nivelul ţesutului adipos şi muscular; Km 5 mmol, valoare apropiată de cea a glicemiei; sinteza şi afinitatea lor")
16
Reglarea exprimării şi afinităţii transportatorilor: Este asigurată de insulină; Este asigurată de insulină; Sensibilitatea la insulină este variabilă în dependenţă de ţesuturi: Sensibilitatea la insulină este variabilă în dependenţă de ţesuturi: în ficat GluT 2 sunt numeroşi şi aparent independenţi faţă de concentraţia insulinei plasmatice; randamentul funcţionării lor este mărit, încât concentraţiile extra- şi intracelulare ale glucozei se echilibrează aproape instantaneu;

17
în ţesutul adipos şi muscular GluT 4 sunt dependenţi de insulină, care stimulează sinteza şi afinitatea lor pentru glucoză, acesta fiind unul din cele mai importante mecanisme de reglare a metabolismului glucidic, deoarece transportorul intracelular al glucozei constituie etapa limitată de viteză a metabolizării sale în aceste ţesuturi.

18
Patologiile medicale: Malabsorbţia glucidelor cauzată de deficienţele dizaharidazelor de la nivelul marginii de perie a enterocitelor; cel mai frecvent fiind deficitul ereditar al lactazei, manifestat prin intoleranţă la lactoză şi la nou-născuţi prin diaree în urma ingestiei de lapte. Malabsorbţia congenitală a glucozei şi galactozei exprimată prin diaree severă, care poate cauza moartea prin deshidratare. Patologie cauzată de deficitul co-transportatorului glucoză-Na +.

19
Ţesuturile necapabile de lactaţie

20
Glanda mamară în perioada de lactaţie

21
Insulinorezistenţa periferică evidenţiată la nivelul celulelor musculare, unde GluT 4 răspund deficitar la secreţia de insulină, ca urmare membranele celulare devin puţin permiabile pentru glucoză, degradarea ei intracelulară fiind diminuată.

22
Metabolismul glicogenului Glicogenul – principala formă de depozitare a glucozei la mamifere; virtual este prezent în fiecare celulă, fiind mai abundent în muşchi şi ficat. Glicogenul – principala formă de depozitare a glucozei la mamifere; virtual este prezent în fiecare celulă, fiind mai abundent în muşchi şi ficat. În muşchi este depozitat sub formă de granule (β-particule) în citozol şi R.E., în ficat totalitatea de β-particule formează α-particule vizibile la microscopul electronic. În muşchi este depozitat sub formă de granule (β-particule) în citozol şi R.E., în ficat totalitatea de β-particule formează α-particule vizibile la microscopul electronic. Greutatea moleculară medie a glicogenului este de câteva milioane Da ( – resturi de glucoză pe moleculă). Greutatea moleculară medie a glicogenului este de câteva milioane Da ( – resturi de glucoză pe moleculă).

23
Granulele depozitate conţin enzime necesare sintezei, degradării şi reglării acestor căi. Conţinutul glicogenului hepatic este dependent de disponibilitatea glucozei şi a precursorilor în gluconeogeneză. Ficatul este responsabil de menţinerea nivelelor glucozei în sânge. Glicogenul muscular diferă mai puţin la semnalele regimului alimentar, dar este dependent de ritmul contracţiilor musculare.

24
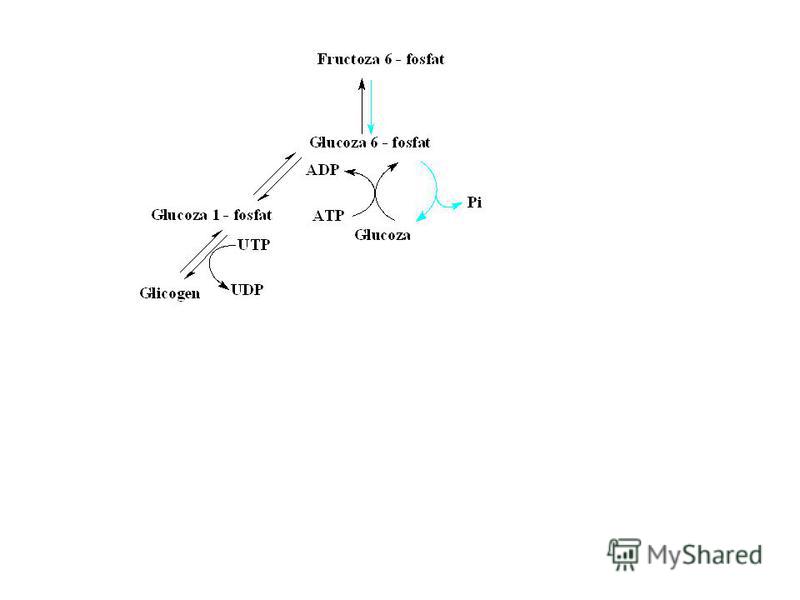
Glicogeneza şi glicogenoliza sunt căi metabolice separate care au numai o enzimă în comun – fosfoglucomutaza. Sinteza şi degradarea glicogenului sunt reglate reciproc – stimularea uneia inhibă alta, viteza sintezei este invers proporţională cu degradarea lui.

25
Mecanismul general de control este fosforilare/defosforilare sau dependent de efectorii alosterici (glucoza, Glu-6-P şi nucleotidele: ATP, ADP, AMP, UDP). Reglarea metabolizmului glicogenului în ficat şi muşchi este diferit, care e dependentă de receptorii respectivi.
. Reglarea metabolizmului glicogenului în ficat şi muşchi este diferit, care e dependentă de rece")
26
Glicogenina Tyr 194 UDP-glucoza UDP Protein-tirozin- glicoziltransferaza (glicogenina) 1
 1")
27
Glicogen sintaza 2 Glicogenina

28
UDP-glucoza UDP 3 Glicogen sintaza

29
UDP-glucoza UDP 4

30
UDP-glucoza UDP Glicogen sintaza, enzima de ramificare a glicogenului 5

31
Particule de glicogen Glicogen sintaza

32
Muşchii: Glicogenul este combustibil pentru metabolismul anaerobic în explozia contracţiei musculare. De altfel şi epinefrina semnalează o necesitate în activitatea musculară. Glicogenul din muşchi nu este o sursă de glucoză pentru alte ţesuturi şi nu e sensibil la nivelul glucozei în sânge. GS musculară (340 kDa) tetramer cu subunităţi identice care există în câteva forme. GS A în forma activă defosforilată conţine cel puţin 9 resturi de serină la extremităţile moleculei care pot fi fosforilate de PK.

33
Terminaţii nereducătoare Legătura (α16) Glicogen Glicogen fosforilaza Molecule de glucoză 1-fosfat Activitate transferazică a enzimei ce hidrolizează structura ramificată Pi
 Glicogen Glicogen fosforilaza Molecule de glucoză 1-fosfat Activitate transferazică a enzimei ce hidrolizează structura ramificată Pi")
34
Activitate (α16) glucozidazică a enzimei ce hidrolizează structura ramificată Glucoză Polimer (α14) neramificat; substrat pentru viitoarea acţiune fosforilazică H2OH2O
 glucozidazică a enzimei ce hidrolizează structura ramificată Glucoză Polimer (α14) neramificat; substrat pentru viitoarea acţiune fosforilazică H2OH2O")
35
GF este reglată de fosforilaz kinaza care convertează forma dimerică prin intermediul Mg 2+ şi fosforilării a două resturi de Ser în forma activă. Fosforilaz kinaza este un hexadecamer care are 4 subunităţi diferite: 1) α (Mm 145 kDa) sau ά (Mm 133 kDa); 2) β (Mm 128 kDa); 3) γ (Mm 44,700 Da); 4) δ (Mm 16,000 Da).
 α (Mm 145 kDa) sau ά (Mm 133 k")
36
Prezenţa subunităţii α sau ά depinde de ţesut; Stoichiometria este α 4 (sau ά 4 )β 4 γ 4 δ 4. Activitatea FK are o cerinţă absolută pentru Ca 2+, care se leagă de subunitatea δ.
β 4 γ 4 δ 4. Activitatea FK are o cerinţă absolută pentru Ca 2+, care se leagă de subunitatea δ.")
37
Secvenţa de aminoacizi a acestei subunităţi este aproape identică cu cea a calmodulinei, cu patru situsuri de legare a calciului. Dar spre deosebire de calmodulină, subunitatea δ este parte integră a enzimei şi nu disociază în absenţa Ca 2+.

38
Metabolismului glicogenului în muşchi

40
Insulina reglează metabolismul glicogenului în muşchi, ultimul este un loc major al absorbţiei glucozei insulino-dependente; ce este mediată de GluT 4. Fixarea insulinei de receptor iniţiază cascada de reacţii fosforilare/defosforilare.

41
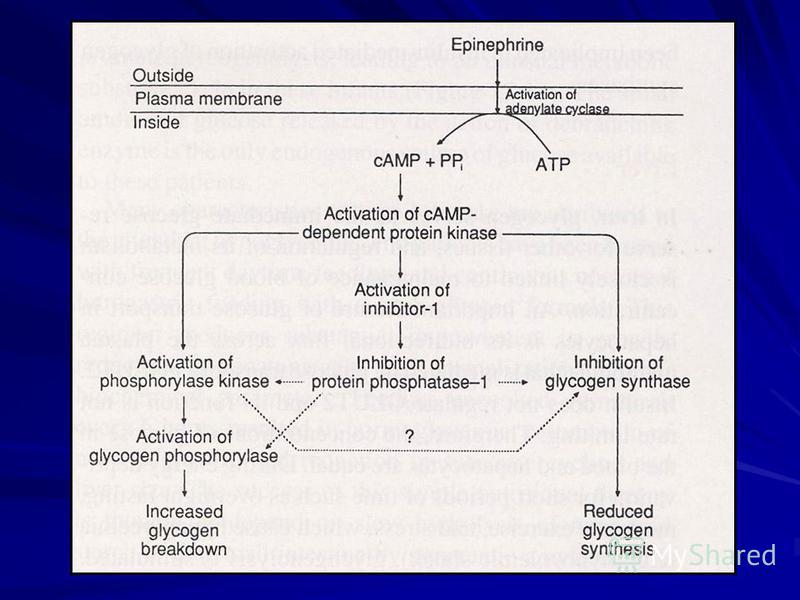
Reglarea metabolismului glicogenului în ficat: El este rezerva imediată de Glu pentru alte ţesuturi – metabolismul cărora este dependent de menţinerea concentraţiei de glucoză în sânge. GluT 2 nu este reglat de insulină – concentraţia Glu în sânge şi hepatocite sunt egale. În perioadele de foame (post în ajun), exerciţii fizice moderate, stresul, şocul hipovolemic este stimulată glicogenoliza.

42
La foame îndelungată gluconeogeneza menţine concentraţia glucozei în sânge. În timpul realimentării rezervele de glicogen sunt restabilite prin G-6-P obţinută în gluconeogeneză (AA, glicerol, propionat). Reglează fenomenele glucagonul, vasopresina, angiotensina II, adrenalina.
. Reglează fenomenele glucagonul, vasopresina, a")
43
Controlul stării fosforilare/defosforilare este mediat de Ca 2+, cAMP, concentraţia glucozei. Creşterea postprandială a Glu în sânge contribuie la inhibarea glicogenolizei şi activarea sintezei.

44
Bolile de depozitare a glicogenului

45
I. Boala von Gierke(glicogenoza hepatorenală): Tip A deficienţă de glucozo-6-fosfatază în ficat, intestin şi rinichi; deficienţă de glucozo-6-fosfatază în ficat, intestin şi rinichi; structura glicogenului este normală; structura glicogenului este normală; hipoglicemie cauzată hormonal, cetoză; hipoglicemie cauzată hormonal, cetoză; galactoza şi fructoza nu sunt convertite la glucoză. galactoza şi fructoza nu sunt convertite la glucoză. Tip B deficienţa transportorului glucozo-6-fosfatului în membrana microzomală a hepatocitului; deficienţa transportorului glucozo-6-fosfatului în membrana microzomală a hepatocitului; structura glicogenului este normală; structura glicogenului este normală; clinic identic cu tipul A. clinic identic cu tipul A.
: Tip A deficienţă de glucozo-6-fosfatază în ficat, intestin şi rinichi; deficienţă de glucozo-6-fosfatază în ficat, intestin şi rinichi; structura glicogenului este normală; structura glicogenului este no")
46
Tip C deficienţa transportului fosfatului în membrana microzomală a hepatocitului; structura glicogenului este normală; clinic identic cu tipul A. Tip D deficienţa transportorului GluT 7 structura glicogenului este normală; clinic identic cu tipul A.

47
II. Boala Pompe (glicogeneză generalizată): deficit de α-1,4-glucozidază lizozomală structura glicogenului este normală; concentraţii excesive de glicogen în vacuole anormale din citozol; în unele cazuri inima este principalul organ implicat cu moarte timpurie, în altele ̶ sistemul nervos este afectat sever; valori normale ale zahărului sanguin.
: deficit de α-1,4-glucozidază lizozomală structura glicogenului este normală; concentraţii excesive de glicogen în vacuole anormale din citozol; în unele cazuri inima este principalul organ implicat cu moart")
48
III. Boala Forbes, Cori (dextrinoza limitată): deficienţa enzimei amilo-1,6-glucozidazei (enzima de deramifiere); structura glicogenului este anormală: lanţul extern lipseşte sau este foarte scurt, numărul punctelor de ramificaţie este mărit; hipoglicemie, răspuns hiperglicemic diminuat la epinefrină sau glucagon şi normal la fructoză şi galactoză; în proces este afectat ficatul, inima, muşchii scheletici.
: deficienţa enzimei amilo-1,6-glucozidazei (enzima de deramifiere); structura glicogenului este anormală: lanţul extern lipseşte sau este foarte scurt, numărul punctelor de ramificaţie este mărit; hipogli")
49
IV. Maladia Andersen (deficienţa de ramificare, amilopectinoză): deficienţa enzimei de ramifiere (1,41,6)- transglucozilazei; deficienţa enzimei de ramifiere (1,41,6)- transglucozilazei; structura glicogenului este anormală: lanţ foarte lung intern şi neramificat extern; structura glicogenului este anormală: lanţ foarte lung intern şi neramificat extern; boală rară, dificil de recunoscut; boală rară, dificil de recunoscut; depozitarea glicogenului anormal; depozitarea glicogenului anormal; progresează ciroza hepatică şi evoluţia este fatală până la vârsta de 20 ani. progresează ciroza hepatică şi evoluţia este fatală până la vârsta de 20 ani.
: deficienţa enzimei de ramifiere (1,41,6)- transglucozilazei; deficienţa enzimei de ramifiere (1,41,6)- transglucozilazei; structura glicogenului este anormală: lanţ foarte lung intern ş")
50
V. Boala McArdle: deficienţa glicogen fosforilazei musculare (miofosforilază); deficienţa glicogen fosforilazei musculare (miofosforilază); structura glicogenului este normală; structura glicogenului este normală; conţinut mărit a glicogenului muscular (2,5- 4,1%, în normă 0,2-0,9%); conţinut mărit a glicogenului muscular (2,5- 4,1%, în normă 0,2-0,9%); nivel scăzut în sânge a lactatului şi piruvatului după exerciţii, scăderea pH-ului nu are loc. nivel scăzut în sânge a lactatului şi piruvatului după exerciţii, scăderea pH-ului nu are loc.
; deficienţa glicogen fosforilazei musculare (miofosforilază); structura glicogenului este normală; structura glicogenului este normală; conţinut mărit a glicogenului muscul")
51
mioglobinurie după exerciţii intense, slăbiciune temporară şi crampe ale muşchilor scheletali; mioglobinurie după exerciţii intense, slăbiciune temporară şi crampe ale muşchilor scheletali; răspuns hiperglicemic normal la epinefrină (datorită enzimei hepatice normale); răspuns hiperglicemic normal la epinefrină (datorită enzimei hepatice normale); pronostic bun. pronostic bun.

52
VI. Boala Hers: deficienţa glicogen fosforilazei hepatice (hepatofosforilaza); deficienţa glicogen fosforilazei hepatice (hepatofosforilaza); structura glicogenului este normală; structura glicogenului este normală; este o formă atenuată a afecţiunii von Gierke; este o formă atenuată a afecţiunii von Gierke; hepatomegalie glicogenică; hepatomegalie glicogenică; hipoglicemie şi cetoză blândă. hipoglicemie şi cetoză blândă.
; deficienţa glicogen fosforilazei hepatice (hepatofosforilaza); structura glicogenului este normală; structura glicogenului este normală; este o formă atenuată a afecţiunii")
53
VII. Maladia Tarui: insuficienţa fosfofructokinazei musculare; insuficienţa fosfofructokinazei musculare; structura glicogenului este normală; structura glicogenului este normală; tablou clinic similar cu maladia McArdle; tablou clinic similar cu maladia McArdle; sunt afectate şi eritrocitele ce determină o hemoliză intensă; sunt afectate şi eritrocitele ce determină o hemoliză intensă; are loc acumularea lactatului; are loc acumularea lactatului; nu este complet clar de ce acest defect rezultă prin creşterea depozitării glicogenului. nu este complet clar de ce acest defect rezultă prin creşterea depozitării glicogenului.

54
VIII. Glicogenoza de tip VIII: reducerea activării fosforilazei în hepatocite şi leucocite; reducerea activării fosforilazei în hepatocite şi leucocite; structura glicogenului este normală; structura glicogenului este normală; hepatomegalie, creşterea depozitării glicogenului hepatic; hepatomegalie, creşterea depozitării glicogenului hepatic; etiologie neclară. etiologie neclară.

55
Deficitul de glicogen sintază se caracterizează prin: hipoglicemie, amplificarea cetogenezei, retard de creştere, deces precoce; se caracterizează prin: hipoglicemie, amplificarea cetogenezei, retard de creştere, deces precoce; Deficitul de fosforilaz kinază (glicogenoză de tip IX) şi deficitul de proteinkinază (glicogenoza de tip X) se caracterizează prin hipoglicemie şi hepatomegalie. se caracterizează prin hipoglicemie şi hepatomegalie.

56
Majoritatea glicogenozelor sunt afecţiuni ereditare ce duc la acumularea glicogenului în ţesuturi şi afectarea metabolismului glucidic, cu generarea simptomelor clinice ca: hepatomegalie, hipoglicemie, hipotonie musculară, deficit energetic în caz de efort fizic. Majoritatea glicogenozelor sunt afecţiuni ereditare ce duc la acumularea glicogenului în ţesuturi şi afectarea metabolismului glucidic, cu generarea simptomelor clinice ca: hepatomegalie, hipoglicemie, hipotonie musculară, deficit energetic în caz de efort fizic.

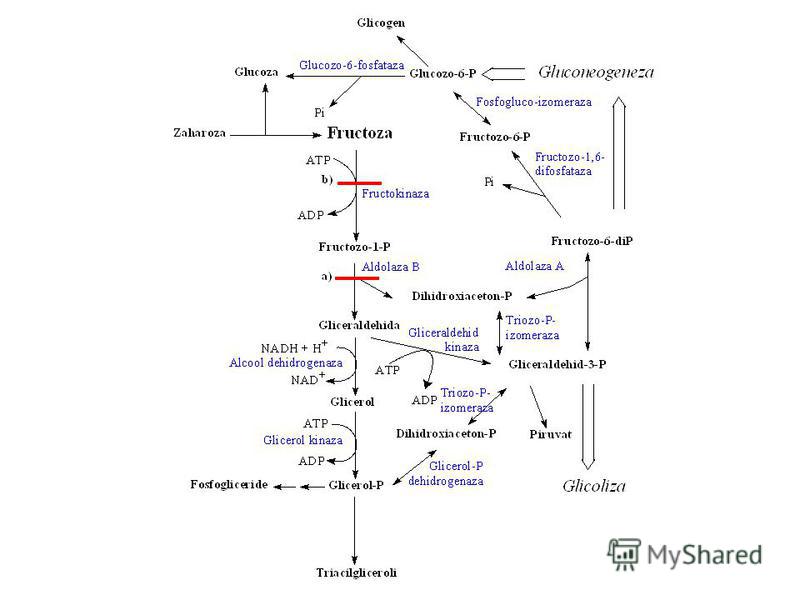
57
Glucoza Glucoza – 6 – fosfat Fructoza – 6 – fosfat Glicoliza (Calea Embden-Mejerhof)
")
58
Fructoza – 6 – fosfat Fructoza – 1,6 – bifosfat Gliceraldehida – 3 – fosfat + Dihidroxiaceton fosfat

59
Gliceraldehid 3 – fosfat (2) 1,3 – Bifosfoglicerat (2) 3 – Fosfoglicerat (2)
 1,3 – Bifosfoglicerat (2) 3 – Fosfoglicerat (2)")
60
2 – Fosfoglicerat (2) Fosfoenolpiruvat (2) Piruvat
 Fosfoenolpiruvat (2) Piruvat")
61
Patologiile medicale: Mutaţiile la nivelul genei glucokinazei cauzează apariţia unei boli monogenice cu debut precoce de tip MODY-2 – scade nivelul glicolizei în ficat şi pancreas ce duce la creşterea glicemiei şi hipersecreţia de insulină.

62
Dar totuşi nivelul insulinei sintetizată este mult diminuat ca urmare a deficitului energetic la nivelul beta celulelor din pancreas. În ficat se diminuează glicogenogeneza şi se amplifică gluconeogeneza, ca răspuns la micşorarea glicolizei.

63
Deficitul piruvat kinazei: Blochează fluxul intermediarilor la nivelul glicolizei. Clinic apar anemii hemolitice ereditare, datorită imposibilităţii desfăşurării glicolizei în eritrocite.

64
Enolaza: Este inhibată sub acţiunea florurei de sodiu, care acţionează ca inhibitor competitiv, blocând glicoliza şi producerea acidului lactic în hematii. Efectul inhibitor se datorează formării unui complex între P, Mg 2+, NaF.

65
Deficitul piruvat dehidrogenazei sau blocarea lanţului respirator mitocondrial: Antrenează acumularea excesivă a acidului piruvic cu geneză glicolitică, care va fi convertit în acid lactic – cu apariţia acidozei respective, primară sau secundară.

66
Deficitul ereditar a LDH: Cauzează apariţia unor miopatii metabolice, manifestate prin reducerea nivelului seric al lactatului şi intoleranţă la efort fizic.

67
Distribuţia izoenzimelor tisulare ale LDH: LDH1 şi LDH2 sunt principalele izoforme în inimă, rinichi, creier şi eritrocite. LDH3 şi LDH4 – predominante în glandele endocrine, splină, timus, leucocite şi trombocite; LDH4 şi LDH5 – preponderent se află în ficat şi muşchii scheletici.

68
Distribuţia izoenzimelor LDH în serul sanguim este următoarea: LDH 1 LDH 3 > LDH 4 LDH 5 Infarctul miocardic: Creşte atât LDH 1 cât şi LDH 2, dar LDH 1 > LDH 2. Maladiile mioplastice, limfoproliferative şi trombocitare: Creşte LDH 3 cu mult faţă de LDH 2.

69
Infarctul pulmonar: Se majorează LDH 2 şi LDH 3. Afecţiuni ale ficatului şi muşchilor: Se depisteză valori majore ale LDH 5.

72
Acidoza lactică primară: Sindrom metabolic caracterizat prin acumularea acidului lactic în sânge (valori mai mari de 5 mmol/L), concomitent cu scăderea pH-ului sanguin sub 7.2.
, concomitent cu scăderea pH-ului sanguin sub 7.2.")
73
Cauzele apariţiei acidozei lactice: deficit ereditar al PDH sau carenţa vitaminei B 1 ; deficit ereditar al PDH sau carenţa vitaminei B 1 ; imposibilitatea regenerării formei oxidate a coenzimei NAD + la nivelul LRmt prin dificitul unei componente sau blocarea FO; imposibilitatea regenerării formei oxidate a coenzimei NAD + la nivelul LRmt prin dificitul unei componente sau blocarea FO; producerea excesivă de NADH (intoxicaţie cu alcool); producerea excesivă de NADH (intoxicaţie cu alcool);

74
deficitul enzimelor gluconeogenezei (piruvat carboxilaza, glucozo-6-fosfataza); deficitul enzimelor gluconeogenezei (piruvat carboxilaza, glucozo-6-fosfataza); accelerarea glicolizei anaerobe (efort fizic prelungit); accelerarea glicolizei anaerobe (efort fizic prelungit);
; deficitul enzimelor gluconeogenezei (piruvat carboxilaza, glucozo-6-fosfataza); accelerarea glicolizei anaerobe (efort fizic prelungit); accelerarea glicolizei anaerobe (")
75
Preponderent se manifestă în perioada neonatală, se caracterizează prin deficit energetic tisular în special în organele cu activitate metabolică intensă (insuficienţă hepatorenală, encefalopatie metabolică). Tratamentul vizează administrarea substanţelor nutritive de natură lipidică a căror oxidare este independentă de CPDH.
. Tratamentul vizează administrarea substan")
76
Acidoza lactică secundară: Se întâlneşte în situaţiile caracterizate prin hipoxie sau anoxie prelungită (şoc, hipoperfizie, insuficienţă cardiovasculară). În carenţele de vitamină B 1.
. În carenţele de vitamină B 1.")
77
Tratamentul constă în mărirea ratei de perfuzie tisulară şi administrarea vitaminei B 1. Se depistează în afecţiunile intestinului subţire (malabsorbţie, rezecţie intestinală) – lactatul este rezultatul activităţii bacteriilor anaerobe. Tratamentul – se utilizează antibiotice, se va limita folosirea carbohidraţilor şi este necesară recolonizarea florei bacteriene.
 – lactatul este rezultatul activităţii bacteriilor anaerobe. Tratamentul –")
78
Gluconeogeneza

81
Gluconeogeneza: Glu – combustibil predominant pentru celulele dependente de metabolismul anaerob, lipsite de mitocondrii şi ţesuturi (creier) care în mod normal nu pot folosi alt combustibil metabolic. Creierul nu poate utiliza acizii graşi, deoarece ei sunt fixaţi de albumina serică şi nu trec prin bariera hematoencefalică.
 care în mod normal nu pot folosi alt combustibil metabolic. Creierul nu poate utiliza acizii graşi, deoarece")
82
Corpii cetonici sunt combustibili alternativi. Conţinutul de glicogen în creier este mic în comparaţie cu necesităţile lui – gluconeogeneza este un proces esenţial pentru supravieţuire. Gluconeogeneza micşorată duce la hipoglicemie, cu o daună ireversibilă pentru creier, nivelul de glucoză mai jos decât 2,2 mmol/L la adulţi, şi 1,7 la nou-născuţi reprezintă o hipoglicemie severă.

83
Un nivel scăzut de glucoză este rezultatul: Utilizării glucozei sau producerii ei scăzute, sau a ambelor cauze. Utilizarea mărită apare în hiperinsulinemie secundară. La o însărcinată diabetică, hiperglicemia mamei duce la hiperglicemia fătului, care cauzează hiperinsulinemie neonatală a fătului.

84
Insuficienţa de glucocorticoizi, glucagon sau hormonului de creştere şi a maladiei severe a ficatului pot produce hipoglicemie, graţie micşorării gluconeogenezei. Insuficienţa de glucocorticoizi, glucagon sau hormonului de creştere şi a maladiei severe a ficatului pot produce hipoglicemie, graţie micşorării gluconeogenezei. Consumul de alcool poate fi cauza hipoglicemiei, acetaldehida formată inhibă fosforilarea oxidativă, promovează glicoliza şi inhibă gluconeogeneza; hipoglicemie şi acidoza lactică sunt trăsături caracteristice a alcoolismului cronic. Consumul de alcool poate fi cauza hipoglicemiei, acetaldehida formată inhibă fosforilarea oxidativă, promovează glicoliza şi inhibă gluconeogeneza; hipoglicemie şi acidoza lactică sunt trăsături caracteristice a alcoolismului cronic.

85
Hipoglicemia intermitentă apare la deficienţa carboxilării piruvatului însoţită de cetoză, acidoză lactică, retard psihomotor sever. Hipoglicemia intermitentă apare la deficienţa carboxilării piruvatului însoţită de cetoză, acidoză lactică, retard psihomotor sever. Deficienţa izoenzimelor citozolice sau mitocondriale ale PEPCK este caracterizată prin inhibarea gluconeogenezei, ambele boli sunt rare, iar deficienţa izoenzimei mitocondriale este moştenită autosomal recesiv. Deficienţa izoenzimelor citozolice sau mitocondriale ale PEPCK este caracterizată prin inhibarea gluconeogenezei, ambele boli sunt rare, iar deficienţa izoenzimei mitocondriale este moştenită autosomal recesiv.

86
Clinic se depisteză hipoglicemie, acidoză lactică, hipotonie, hepatomegalie. Clinic se depisteză hipoglicemie, acidoză lactică, hipotonie, hepatomegalie. Tratamentul este simptomatic, de susţinere. Tratamentul este simptomatic, de susţinere. Deficienţa F-1,6-DP moştenită autosomal recesiv afectează sever gluconeogeneza cauzând hipoglicemie, cetoză şi acidoză lactică. Deficienţa F-1,6-DP moştenită autosomal recesiv afectează sever gluconeogeneza cauzând hipoglicemie, cetoză şi acidoză lactică.

87
Deficitul G-6-P-aza, enzimă comună gluconeogenezei şi glicogenolizei este responsabilă de boala von Gierke. Deficitul G-6-P-aza, enzimă comună gluconeogenezei şi glicogenolizei este responsabilă de boala von Gierke. Acumularea de G-6-P are ca consecinţă sinteza excesivă a glicogenului în ficat şi rinichi, activarea şuntului pentozofosfat cu producerea masivă de R-5-P, şi implicit de nucleotide purinice având în consecinţă hiperuricemia.

88
Hipoglicina A toxina unui fruct verde din Jamaica, inhibă gluconeogeneza, provoacă hipoglicemie; formează esteri nemetabolizabili cu CoA şi carnitină – inhibând oxidarea acizilor graşi. Ca urmare se formează acidoza lactică manifestată prin tulburări neurologice, digestive şi musculare.

89
Reglarea hormonală a nivelului glucozei în sânge: Un rol unic îi aparţine insulinei – hormon ce facilitează depozitarea tuturor tipurilor de bază ale substanţelor energetice. Este unicul hormon secreţia căruia este dependentă de nivelul glucozei sanguine. Toţi ceilalţi hormoni necesită ca excitanţi o hipoglicemie vădită sau intercalarea semnalelor de tipul stresului, raţiei alimentare proteice, efortului fizic.

90
Adrenalina, glucagonul, cortizolul, somatotropina, tiroxina – accelerează utilizarea de energie, măresc nivelul glucozei prin următoarele mecanisme: glucagonul amplifică glicogenoliza şi gluconeogeneza; glucagonul amplifică glicogenoliza şi gluconeogeneza; cortizolul facilitează gluconeogeneza şi blochează absorbţia glucozei; cortizolul facilitează gluconeogeneza şi blochează absorbţia glucozei;

91
catecolaminele favorizeză glicogenoliza şi blocheză absorbţia glucozei; catecolaminele favorizeză glicogenoliza şi blocheză absorbţia glucozei; somatotropina inhibă absorbţia glucozei; somatotropina inhibă absorbţia glucozei;

92
Patologii medicale: Insulinorezistenţa periferică – sindrom biochimic caracterizat prin creşterea marcată a sintezei de insulină în pofida unui nivel normal al glicemiei, cauzat de unele mutaţii ale genei ce codifică receptorul de insulină; apare imposibilitatea transmiterii intracelulare a mesajului hormonal.

93
Această categorie include diverse afecţiuni ereditare cum ar fi: diabetul lipoatrofic infantil, diferite forme de obezitate, sindromul Donahue (dismorfism facial, caşexie, retard mintal, hipotrofie staturală) sau diverse tulburări endocrine. Această categorie include diverse afecţiuni ereditare cum ar fi: diabetul lipoatrofic infantil, diferite forme de obezitate, sindromul Donahue (dismorfism facial, caşexie, retard mintal, hipotrofie staturală) sau diverse tulburări endocrine. O mutaţie la nivelul situsului de scindare a precursorului insulinei generează creşterea marcată a proinsulinei în circulaţie, în detrimentul insulinei active. O mutaţie la nivelul situsului de scindare a precursorului insulinei generează creşterea marcată a proinsulinei în circulaţie, în detrimentul insulinei active.
 sau diverse tulburări endocrine. Această")
94
Diabetul zaharat – sunt descrise mai multe tipuri: Tip I – insulinodependent: este cauzat de deficitul insulinei, consecinţa unor mecanisme autoimune (este depistată prezenţa anticorpilor anticelulei β); boala debutează timpuriu, se manifestă prin deficitul total a insulinei, concomitent cu degenerarea marcată a celulelor β. Tip II – non-insulinodependent – afecţiune multifactorială, relativ frecventă este asociat frecvent cu obezitate, insulinorezistenţa periferică şi perturbarea metabolismului lipidic.
; boala debutează timpuriu, se manifestă prin de")
95
Tip MODY - 2 – o afecţiune ereditară monogenică provocată de deficitul glucokinazei; se observă diminuarea glicolizei şi a glicogenogenezei în ficat, ceea ce determină activarea gluconeogenezei. Secundar – se desemnează în pancreatite cronice sau în alte afecţiuni care lizează integritatea pancreasului.

96
Secundar cu exces de hormoni hiperglicemici – în afecţiunile endocrine, caracterizate prin hipersecreţia de catecolamine, cortizol, somatotropina; aceste sindroame sunt însoţite de insulinorezistenţă periferică. Gestaţional – debutează pe parcursul sarcinii şi poate evalua spre instalarea diabetului zaharat, reducerea toleranţei la glucoză.

97
Glucozo – 6 – fosfat glucozo – 6 – fosfat dehidrogenaza HMS

98
6 – Fosfo-Glucono- δ-Lactonă lactonază

99
6 – Fosfo-Gluconat 6 – fosfogluconat dehidrogenază

100
D – Ribulozo-5-fosfat D – Ribozo-5-fosfat Fosfopentoz izomerază

101
reacţiile nonoxidative ale căii pentozofosfat

102
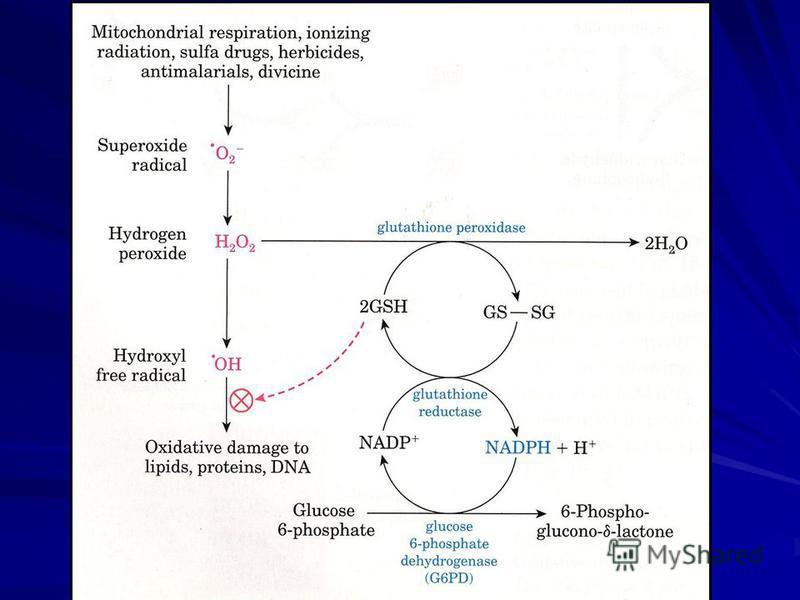
HMS şi celulele roşii ale sângelui: Şuntul este foarte activ în eritrocite. NADPH format protejează AGN de interacţiunea anormală cu O 2 (în membrană) şi asigură gradul de oxidare a Fe 2+ în Hb. La insuficienţa de G-6-P-DH apare o hemoliză patologică, determinată de gradul mic de reducere a glutationului; în consecinţă eritrocitele sunt expuse la efectele nocive ale radicalilor liberi.
 şi asigură gradul de oxidare a Fe 2+ în Hb. La insuficienţa de G-6-P-DH apare o hemoliză patologică")
103
Deosebit de activ este şuntul în celulele fagocitare unde are loc liza bacteriilor şi celulelor anormale. NADPH + 2O 2 2O 2 - +NADP + + H + Enzima este o NADP-oxidază; în fagolizozomi anionul superoxid generează spontan H 2 O 2, substrat pentru mieloperoxidază, prezentă în granulele neutrofile primare, în prezenţa metalelor H 2 O 2 şi anionul superoxid va genera radicalul hidroxil şi oxigenul singlet ( · O 2 ). 2O H + H 2 O 2 + O 2 (SOD) 2O H + H 2 O 2 + O 2 (SOD) O H 2 O 2 · O 2 + OH - + OH ·

104
Radicalul hidroxil (OH) este foarte activ şi va oxida diferite molecule. Şuntul din eritrocite este singura sursă de NADPH. Intră în componenţa glutation reductazei unde electronul se transferă de la NADPH la FAD, apoi la punţile disulfidice în subunitate şi apoi pe glutationul oxidat.
 este foarte activ şi va oxida diferite molecule. Şuntul din eritrocite este singura sursă de NADPH. Intră în componenţa glutation reductazei unde electronul se transferă de la NADPH la FAD, apoi la punţile disulfidice în subun")
105
GR reprezintă un dimer cu o masă moleculară de 50 kDa. Fiecare subunitate este constituită din 3 domenii structurale: fixatori de FAD, NADPH şi domeniul intermediar. Relaţia GSH/G-S-S-G în condiţii normale este 500. GSH joacă un rol deosebit în procesele de detoxicaţie, este utilizat în inactivarea potenţialului de distrugere a peroxizilor organici şi a H 2 O 2, rezultant al acţiunii SOD.

106
GP enzimă seleno-constituentă neutralizează peroxidul: 2GSH+H 2 O 2 G-S-S-G + 2H 2 O. Scăderea nivelului GSH se depistează în favism (deficit congenital de G-6-P-DH) – leziune metabolică, indivizii prezintă o mare sensibilitate la sulfamide sau medicamente antimalarice cu declanşarea unor manifestări hemolitice generalizate.
 – leziune metabolică, indivizii prezintă o mare sensibilitate la sulfamide sau medic")
107
Defectele genetice în activitatea γ-glutamil cistein sintazei şi glutation sintazei sunt cauzele anemiilor hemolitice. De asemenea deficienţile nutriţionale în riboflavină sau seleniu determină anormalităţi în metabolismul glutationului. O ameliorare parţială se obţine la utilizarea vitaminei E şi antioxidanţilor.

109
Acidul glucuronic

110
Acidul glucuronic este utilizat la: Conjugarea diverşilor compuşi endo- şi exogeni din clasa aminelor, fenolilor, acizilor carboxilici. UDP-glucuronatul este donator de glucoronil în sinteza polizaharidelor acide (proteoglicani – glicozoaminglicani). Se conjugă cu hormonii tiroidieni în hipertiroidism. La nivel scăzut de hormoni are loc hidroliza acestor compuşi cu eliberarea hormonilor activi.
.")
111
Reacţiile sunt catalizate de glucuronil transferaze – enzime inductibile. Reacţiile sunt realizate în ficat şi reprezintă o etapă de detoxifiere a compuşilor străini (xenobiotice) sau proprii organismului, formând glucuronide – compuşi cu polaritate evidentă, facilitând transportul şi excreţia lor.
 sau proprii organismului, formând glucuronide – compuşi cu polaritate")
112
Imaturitatea sau deficienţele genetice ale acestor enzime duc la perturbarea proceselor de detoxifiere. Când necesităţile organismului în acid glucuronic sunt satisfăcute, metabolizarea lui conduce la D- xiluloză, substrat al HMS.

113
Maladia ereditară - pentozuria esenţială se datoreză imposibilităţii conversiei L- în D- xiluloză, ca rezultat al absenţei enzimei L- xilitol-DH – NADPd, cu eliminarea în urină a L-xilulozei, conform reacţiilor:

114
La animale, cu excepţia maimuţelor antropoide, cobaiul şi omului, acidul L-gulonic poate fi convertit în acid ascorbic. Incapacitatea de a sintetiza ascorbatul (vitamina C) se datorează lipsei ereditare a gulonolacton-oxidazei.
 se datorează lipsei ereditare a gulonolacton-oxidazei.")
115
UDP-glucoz dehidrogenaza UDP-glucoza UDP-D-glucuronat Inserţia glucoronatului în glicozaminoglicani aşa ca hialuronatul, condroitin sulfatul Glucuronidarea medicamentelor, toxinelor Sinteza vitaminei C

116
D-Glucuronat L-Gulonat L-Gulonolactonă Acidul ascorbic (vitamina C) glucuronat reductaza aldonolactonaza gulonolacton oxidaza
 glucuronat reductaza aldonolactonaza gulonolacton oxidaza")
118
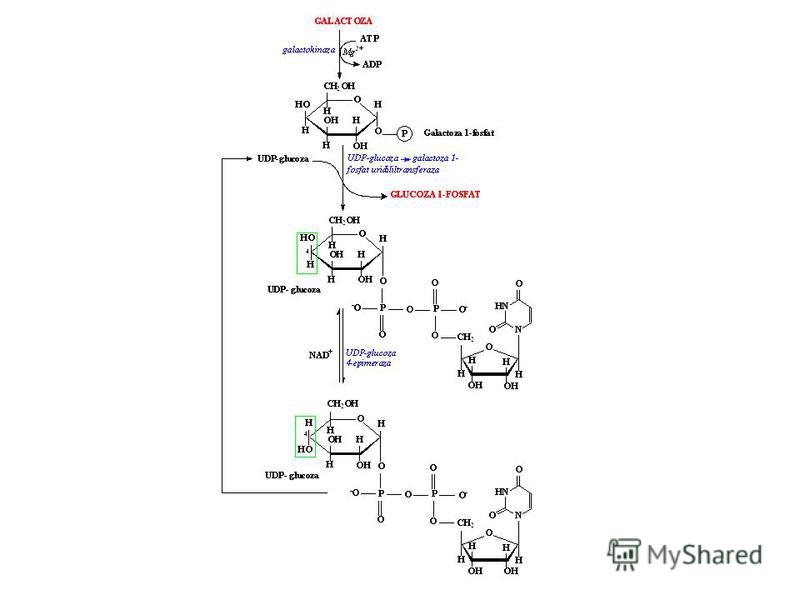
Patologiile medicale: La insuficienţa enzimei UDP-α-glucoză-α- galacto-1-P-uridil-transferaza are loc acumularea în sânge a Gal-1-P; consecinţele sunt nefavorabile pentru organism: mărirea ficatului şi a altor organe, cataracta, dereglări mintale; se mai acumulează şi galactitolul – cauza directă a cataractei; suferă preponderent copiii, de aceea reducând sau eliminând complet laptele din raţie, scad simptomele patologice;

119
Galactozemia şi galactouria sunt provocate de deficienţa galactokinazei care conduce la acumularea galactitolului, dacă în dietă este prezentă galactoza.

120
Aldozreductaza - enzimă prezentă în ficat, ţesutul nervos, veziculele seminale, nu este importantă din punct de vedere fiziologic în metabolismul galactozei dacă nivelul galactozei nu este ridicat; Intoleranţa la lactoză este de 3 tipuri: copii prematuri (deficit congenital); copii prematuri (deficit congenital); deficitul care apare ca rezultat a înlăturării chirurgicale a unei porţiuni din intestinul subţire; deficitul care apare ca rezultat a înlăturării chirurgicale a unei porţiuni din intestinul subţire;

121
deficit provocat de lezarea celulelor mucoase intestinale – care necesită eliminarea din dietă a sursei majore de galactoză; deficit provocat de lezarea celulelor mucoase intestinale – care necesită eliminarea din dietă a sursei majore de galactoză; Organismul poate sintetiza cantitatea suficientă de UDP-Gal prin reacţia de epimerizare.

123
Patologii medicale: Intoleranţa ereditară la fructoză – se caracterizează prin absenţa aldolazei B, care conduce la acumularea intracelulară a F-1-P. Ultima inhibă Glu-6-P-aza şi glicogen fosforilaza şi prin urmare glucoza este depozitată în ficat ca ester fosforic, ceea ce explică hipoglicemia severă, voma, icterul şi hemoragia; poate provoca insuficienţa hepatică.

124
Fructozuria este provocată de deficienţa ereditară a fructokinazei, care conduce la acumularea fructozei în sânge. Deficitul enzimelor responsabile de interconversia monozaharidelor şi de biosinteza glicoproteinelor, se exprimă prin sindromul deficienţei carbohidraţilor din structura glicoproteinelor. Afecţiuni ereditare care se manifestă prin retard de creştere, anomalii neurologice şi afectare multienzimatică.

125
Diagnosticul de laborator constă în analiza profilului electroforetic al transferinei serice, care migrează mai lent în absenţa acidului sialic din componenta lanţurilor oligozaharidice ale proteinei. Diagnosticul de laborator constă în analiza profilului electroforetic al transferinei serice, care migrează mai lent în absenţa acidului sialic din componenta lanţurilor oligozaharidice ale proteinei.

126
O grupă de glicoproteine ce conţin acid sialic sunt prezente în saliva oamenilor sănătoşi, la pierderea acidului sialic, graţie unor enzime salivare, aceste proteine precipită pe suprafaţa smalţului dentar, generând placa dentară - cauza cariei şi maladiei paradontale; În condiţii normale acidul sialic nu precipită şi prin urmare glicoproteinele salivare protejează dinţii de diferite afecţiuni.

127
În salivă se întâlnesc şi polizaharide extracelulare de origine bacteriană: dextranii – polimeri ai glucozei (α-1,6; β-1-3 şi 1-2) şi levanii (polimeri ai fructozei – 1,2); aceşti polimeri sunt adevăraţi lianţi ai plăcii dentare, se formează pe contul zaharozei, considerată, din acest motiv, cea mai cariogenă.
 şi levanii (polimeri ai fructozei – 1,2); aceşti polimeri sunt adevăraţi lianţi ai plăcii dentare, se formează pe contu")
128
În natură ne mai întâlnim cu un diglucid – amigdalina, care se află în migdalele amare, în sâmburi de piersici, caise, prune. La hidroliza ei formează două molecule de β–D–glucoză, una de aldehidă benzoică şi una de acid cianhidric (HCN).
.")
129
Enzimele florei intestinale sunt capabile să hidrolizeze amigdalina, cu eliberarea acidului cianhidric; absorbţia lui generează efectul toxic. Amigdalina se consideră agent antitumoral. Tumorile posedă activitate β-glucuronidazică sau β-glucozidazică eliberând HCN – cauza morţii celulare. Amigdalina se consideră agent antitumoral. Tumorile posedă activitate β-glucuronidazică sau β-glucozidazică eliberând HCN – cauza morţii celulare.

Еще похожие презентации в нашем архиве:




, dizaharide (30)% şi monozaharide. În alimentaţie glucidele.")



 SEDIUL ACTIVITĂŢILOR METABOLICE SEDIUL ACTIVITĂŢILOR METABOLICE DIAMETRU : μ m mm DIAMETRU.")


 Starea solidă: au formă proprie; rigiditate; au volum propriu (incompresibilitate). b) Starea lichidă:")



 - 1953 - E.C.S. Slater.")







